
ВВЕДЕНИЕ
Сердечно-сосудистые заболевания (ССЗ) остаются ведущей причиной смерти во всем мире [1, 2]. Измерение биологических маркеров произвело революцию в диагностике и контроле за эффективностью проводимого лечения пациентов с CCЗ. К наиболее широко используемым современным биомаркерам относятся натрийуретические пептиды и сердечные тропонины. Было выявлено и множество других биомаркеров, но лишь немногие из них нашли применение в реальной клинической практике [3, 4].
Представленные нами данные посвящены транскрипционно-модулирующему фактору Jun dimerization protein 2 (JDP2) и его роли при кардиоваскулярной патологии.
МЕТОДОЛОГИЯ ПОИСКА ИСТОЧНИКОВ
В статье представлен обзор актуальных публикаций, посвященных изучению JDP2. Анализ источников литературы проводился в базах данных PubMed, РИНЦ, MedLine, Google Scholar, Science Direct. Рассматривались зарубежные и отечественные публикации. Поиск выполнялся соответственно по следующим ключевым словам: JDP2, «биологические маркеры», «сердечно-сосудистые заболевания», biological markers, cardiovascular disease. Результаты различных исследований свидетельствуют, что существует научный интерес к изучению диагностической и прогностической ценности JDP2 при кардиоваскулярных заболеваниях.
БИОЛОГИЧЕСКИЕ АСПЕКТЫ JDP2
Белок димеризации JDP2 представляет собой фактор транскрипции bZip-типа, который принадлежит к семейству активаторных белков-1 (AP-1) [5, 6]. JDP2 первоначально был описан как репрессор AP-1 [5]. Он угнетает 12-O-тетрадеканоилфорбол-13-ацета-зависимую транскрипцию или аденозинмонофосфат зависимую транскрипцию путем гетеродимеризации с белками семейства Jun (Jun-C и Jun-B) [7]. JDP2 эпигенетически подавляет экспрессию генов за счет рекрутирования гистоновых деацетилаз, таких как, например, гистондеацетилаза-3 [8].
В 2013 г. Tanigawa S. et al. доказали, что JDP2 выступает в роли коактиватора рецептора прогестерона и фактора 2, связанного с транскрипционным фактором NF-E2 (Nrf2) [9]. У мышей с нокаутом JDP2 обнаружена аномальная дифференцировка остеокластов и нейтрофилов [10]. Nakade K. et al. опубликовали данные о том, что JDP2 подавляет дифференцировку адипоцитов [11]. Было показано, что активация фактора транскрипции 4 (ATF4) регулирует экспрессию гена JDP2 [12, 13]. Однако точный механизм, с помощью которого ATF4 активирует экспрессию JDP2, остается неизвестным [14].
Совсем недавно повышенная регуляция длинной некодирующей рибонуклеиновой кислоты (РНК) TTTY15 была обнаружена при инфаркте миокарда (ИМ) в кардиомиоцитах человека в условиях гипоксии [5]. TTTY15 нацелен на микроРНК-455, которая регулирует экспрессию JDP2. Таким образом, существуют доказательства индукции JDP2 в условиях гипоксии в кардиомиоцитах человека [5].
JDP2 И СЕРДЕЧНО-СОСУДИСТАЯ ПАТОЛОГИЯ
Первый отчет о связи повышенной экспрессии JDP2 после острого ИМ (ОИМ) с прогрессированием сердечной недостаточности (СН) был опубликован Maciejak A. et al. в 2015 г. [15]. Ученые собрали образцы периферической крови у 111 пациентов с ИМ с подъемом сегмента ST (ИМпST); контрольную группу составил 41 пациент со стабильной ишемической болезнью сердца (ИБС) и без ИМ в анамнезе. На основании уровня N-концевого фрагмента мозгового натрийуретического гормона В-типа (NT-proBNP) в плазме крови и значений фракции выброса левого желудочка (ФВ ЛЖ) пациенты с ИМпST были разделены на 2 группы: с СН и без нее. Было обнаружено, что экспрессия JDP2 при ОИМ отмечена у больных с большей частотой развития СН после ОИМ. Это свидетельствует в пользу того, что JDP2 может служить ценным лабораторным маркером для прогнозирования развития СН у пациентов с ИМ [15].
В 2019 г. было показано, что JDP2 принимает активное участие в белок-белковом взаимодействии (protein–protein interactions, PPIs) у пациентов с ИМ [16].
Kehat I. et al. оценивали экспрессию JDP2 и ее связь с развитием дилатации полостей сердца на экспериментальных моделях 4-месячных мышей. Экспрессия JDP2 привела к массивной дилатации предсердий, нарушениям проводимости сердца и летальному исходу [17].
Shiai. W. и Na C. опубликовали данные исследования, посвященного изучению прогностической роли JDP2 в развитии СН после перенесенного ИМ [18]. Было идентифицировано несколько пар регуляторных взаимодействий между микроРНК и информационными РНК, в том числе hsa – miR – 17‑3p – JDP2. Эти результаты нашли подтверждение при обследовании небольшой группы пациентов с ИМ, у которых было обнаружено понижение регуляции has-mir-17-3p и повышение регуляции микроРНК JDP2 в крови [18].
Немецкими и венгерскими учеными было выполнено исследование in vivo на мышах со сверхэкспрессией JDP2 с целью изучения влияния JDP2 на прогрессирование СН. С момента рождения до возраста 4 нед избыточная экспрессия JDP2 предотвращалась путем кормления трансгенных мышей доксициклином, далее происходила сверхэкспрессия JDP2. Уже через 1 нед показатели сердца по данным эхокардиографии (ЭхоКГ) ухудшились. Через 5 нед было отмечено падение артериального давления (АД), ФВ ЛЖ и формирование дилатации ЛЖ. Также было обнаружено увеличение массы сердца, экспрессии микроРНК предсердного натрийуретического пептида (ANP), маркеров воспаления, коллагена и фибронектина. Экспрессия белка коллагена 1 увеличилась, вследствие чего развился фиброз. Критерием повышенного ремоделирования внеклеточного матрикса явилось увеличение активности матриксной металлопротеиназы 2 (MMP2, MMP-2; желатиназа A) [19].
Heger J. et al. сообщили о сократительной дисфункции и на клеточном уровне: выделение кардиомиоцитов желудочков у мышей выявило снижение скорости укорочения и сокращения клеток при электростимуляции после одной недели сверхэкспрессии JDP2. После продолжительной сверхэкспрессии JDP2 сократительная способность снижалась еще больше, а после пожизненной сверхэкспрессии JDP2 положительные инотропные эффекты за счет бета-адренергической стимуляции нивелировались [20]. Также было описано нарушение кальциевого насоса саркоплазматического ретикулума (SERCA) в желудочках взрослых мышей со сверхэкспрессией JDP2 [19] (рис. 1).
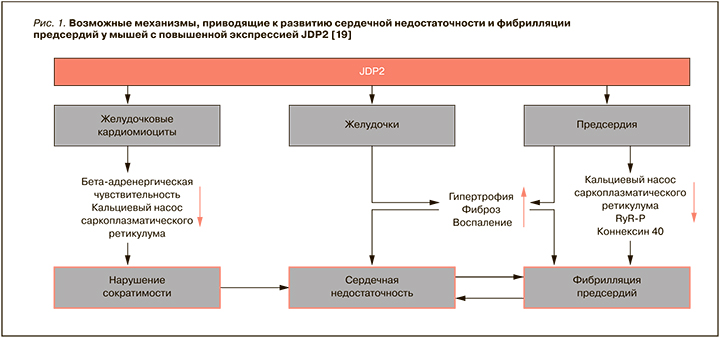
Представляет интерес и исследование, проведенное Koren L. et al. Ученые опубликовали данные о том, что у мышей, чрезмерно экспрессирующих гомолог JDP2, активирующий фактор транскрипции 3 (ATF3), развивается увеличение массы сердца и нарушение его функции [21]. На моделях трансгенных мышей с сердечной сверхэкспрессией ATF3 было показано дезадаптивное ремоделирование сердца и снижение его функции. Также было обнаружено, что экспрессия ATF3 индуцируется сердечной ишемией в сочетании с реперфузией (ишемия–реперфузия) как в культивируемых клетках, так и в моделях на животных. У трансгенных мышей, экспрессирующих ATF3, обнаружили расширение и гипертрофию предсердий и желудочков. Микроскопическое исследование продемонстрировало дегенерацию и фиброз миоцитов. Трансгенное сердце имело пониженную сократимость и аберрантную проводимость. Интересно, что экспрессия белка сорцина, ингибирующего высвобождение кальция из саркоплазматического ретикулума, была повышена в сердцах трансгенных мышц. Эти результаты показали, что экспрессия ATF3 приводит к изменению экспрессии генов и нарушению функции сердца.
Предполагается, что ATF3 и JDP2 являются близкими структурными гомологами с похожими целевыми последовательностями в промоторе различных генов, которые дают аналогичные эффекты при их активации. Также было описано, что у мышей с двойным дефицитом JDP2 и ATF3 наблюдалось снижение дезадаптивного ремоделирования сердца и более низкая гипертрофия после продольного сужения аорты. Таким образом, баланс JDP2 и ATF3, по-видимому, служит весовым фактором в развитии СН [21, 22].
Как уже отмечено выше, описание трансгенных мышей JDP2 с непрерывной кардиоспецифической сверхэкспрессией JDP2 от рождения до 4-недельного возраста позиционировало JDP2 в качестве основного медиатора выраженной дилатации предсердий [17]. Показательно, что аритмии и дилатация предсердий были почти полностью обратимы при прекращении сверхэкспрессии JDP2 [17].
Parahuleva M. et al. выполнили исследование in vivo на мышах со сверхэкспрессией JDP2 для изучения влияния JDP2 на предрасположенность к спонтанной фибрилляции предсердий (ФП). Спонтанное начало ФП было зарегистрировано с помощью электрокардиограммы (ЭКГ) в сроки 4–5 нед сверхэкспрессии JDP2. В предсердной ткани мышей JDP2, помимо 3,6-кратного увеличения микроРНК JDP2, не удалось обнаружить никаких изменений в течение одной недели сверхэкспрессии JDP2. Дилатация и гипертрофия предсердий, удлинение кардиомиоцитов и фиброз стали очевидными через 5 нед сверхэкспрессии JDP2. На записи ЭКГ были выявлены удлинение интервала PQ и расширение зубцов P и QRS-комплексов, а также атриовентрикулярная блокада и пароксизмальная форма ФП. Наряду с этим было обнаружено снижение уровней белков NCX, Cav1.2 и RyR2, транспортирующих ионы кальция, а также мРНК коннексина 40. Также было отмечено увеличение уровней ANP, моноцитарного хемотаксического белка 1 (MCP1), маркеров инфильтрации иммунных клеток (CD68, CD20) у мышей со сверхэкспрессией JDP2 [23].
Kehat I. et al. опубликовали данные о том, что сердечная сверхэкспрессия нескольких факторов, подавляющих транскрипцию членов семейства bZIP, таких как нефосфорилируемая форма транскрипционного фактора CREB и ATF3, вызывала массивную дилатацию предсердий. Чтобы попытаться охарактеризовать этот путь, ученые выделили мощный ингибирующий bZIP белок димеризации Jun 2. Экспрессия JDP2 влекла за собой массивную дилатацию предсердий, потерю экспрессии коннексина 40 (Cx40), коннексина 43 (Cx43) и легкой цепи миозина 2 (MLC2a), а также нарушения атриовентрикулярной проводимости и летальный исход. Ингибирование экспресии JDP2 нивелировало его негативное влияние на сердце. Авторы заключили, что ингибирование bZIP может стать новой лекарственной мишенью для предотвращения дилатации предсердий [24].
Huang S. et al. описали повышенную экспрессию TTTY15, ассоциированную с гипоксией, в кардиомиоцитах пациентов с ИМ. Было показано, что подавление длинной некодирующей РНК TTTY15 ослабляет индуцированное гипоксией повреждение кардиомиоцитов [25].
Резюме доступных клинических исследований, которые описывают эффекты JDP2, связанные с кардиологическими заболеваниями, представлено в таблице.
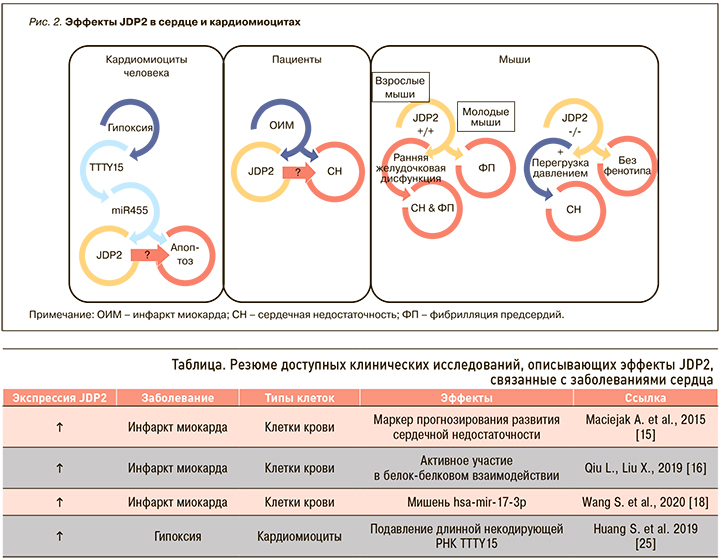
На рисунке 2 обобщены результаты имеющихся данных о кардиальных эффектах JDP2.
ЗАКЛЮЧЕНИЕ
Определение новых сердечно-сосудистых биомаркеров, анализ их патофизиологических аспектов и изменения концентрации под влиянием различных вариантов лечения позволяют нам понять многие патогенетические особенности развития и течения СН [26, 27]. На сегодня определение концентраций BNP и NT-proBNP служит неким «золотым стандартом» диагностики СН и прогнозирования ее течения, однако ограничения, обусловленные влиянием многих факторов на их концентрации, неопределенность пороговых значений и низкая информативность при СН с нормальной ФВ ЛЖ предопределяют необходимость дальнейшего поиска высокочувствительных и специфичных лабораторных маркеров [3, 4, 28]. Новые маркеры, такие как маркер фиброза галектин-3 (Gal-3), стимулирующий фактор роста ST2, пептидный гормон адреномедуллин, суррогатный маркер вазопрессин, хемокин-CX3CL1 и другое, все больше находят свое место в клинической практике [3, 4, 29]. Кроме того, весьма важен мультимаркерный подход к диагностике СН, стратификации ее риска и оценке эффективности лечения [3, 4, 30].
Появляются работы, позволяющие рассматривать роль JDP 2в диагностике и оценке прогноза у пациентов кардиологического профиля. Исследования на трансгенных грызунах и ряд клинических исследований показали, что JDP2 принимает участие в развитии ФП и СН (см. рис. 1). Тем не менее точная функция этого биологического маркера еще достаточно туманна, что предопределяет необходимость будущих научных и клинических исследований в этом направлении.


