
ВВЕДЕНИЕ
Болезнь Вильсона–Коновалова (болезнь Вильсона, гепатоцеребральная дистрофия, гепатолентикулярная дегенерация) представляет собой тяжелое аутосомно-рецессивное наследственное заболевание с хроническим прогрессирующим течением, характеризующееся избыточным накоплением в организме меди и ее и токсическим воздействием. Болезнь сопровождается сочетанным поражением паренхиматозных органов (прежде всего печени) и головного мозга [1].
ЭПИДЕМИОЛОГИЯ
Болезнь Вильсона–Коновалова (БВК) распространена повсеместно с частотой от 1–5 на 30–300 тыс. населения; при гетерозиготном носительстве патологического гена этот показатель варьирует от 1:90 до 1:224 [2, 3]. Стабильно высокая частота БВК фиксируется в регионах, где распространены близкородственные браки (Иран, Йемен, Иордания, Япония, Индия, Китай, южные регионы Италии) [4–7]. В России встречаемость заболевания находится в пределах 0,3:100 000–1,8:100 000 населения [8]. БВК с одинаковой частотой выявляется как у мужчин, так и женщин и отмечается в семьях, где оба родителя являются гетерозиготными носителями мутантного гена, но клинически здоровыми лицами без аналогичного заболевания в своей родословной. Заболевают БВК только гомозиготные или компаунд-гетерозиготные носители мутаций гена АТР7В [9]. Имеются данные о конкордантности по БВК у однояйцевых близнецов.
Манифестировать заболевание может в любом возрасте, средний возраст ее манифестации – 11–25 лет [10]. С возрастом частота встречаемости этой патологии снижается. На долю лиц старше 40 лет с хроническим активным гепатитом неясной этиологии приходится 3% пациентов с БВК [11]. Тем не менее в литературе приводятся случаи диагностированного цирроза печени, обусловленного БВК, у пациентов в возрасте 8 мес, 3 лет [12] и 80 лет [13, 14]. Начавшись в более позднем возрасте, болезнь протекает медленнее, тогда как ее дебют в молодом возрасте без назначения адекватного лечения характеризуется быстрым развитием необратимых изменений с фатальным исходом [10].
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Причиной БВК служат мутации гена ATP7B на длинном плече 13 хромосомы (13q14.3–q21.1), которая кодирует транспортирующую медь АТФазу P-типа и отвечает за внутриклеточный транспорт меди [15]. В настоящее время известно более 900 мутаций в гене АТР7В, из которых 380 имеют подтвержденную роль в патогенезе заболевания. Наиболее распространенной мутацией в Европе и Северной Америке является H1069Q [16]. Мутация R778L [17, 18] встречается в Азии и определяется у 57% пациентов моложе 18 лет. В Сардинии зарегистрирована редкая и характерная только для этого региона мутация – del 1441-427 (66,5%) [19, 20]. В России наиболее частыми оказались мутация H714G и миссенс-мутация His1069Gln [8]. Помимо мутаций, характерных для определенной этнической группы, существует спектр мутаций de novo, который постоянно расширяется.
ПАТОГЕНЕЗ
Мутация гена АТР7В обусловливает нарушение процесса включения меди в апоцерулоплазмин с последующим снижением синтеза церулоплазмина [21]. Недостаточная экскреция меди с желчью приводит к токсическому ее депонированию в гепатоцитах, накоплению свободных радикалов и активации процессов перекисного окисления липидов. Это стимулирует синтез коллагена в гепатоцитах с последующим развитием жировой дистрофии, воспалительной реакции в области портальных трактов, некроза гепатоцитов и формированием мелкоузлового цирроза печени.
Свободная медь, высвободившаяся из поврежденных гепатоцитов и не связанная с церулоплазмином, поступает в избыточном количестве в кровеносное русло, вызывая повреждение мембран и ферментных систем эритроцитов с развитием Кумбс-отрицательного внутрисосудистого гемолиза. Накопление меди в структурах головного мозга (в зубчатых и чечевицеобразном ядрах, хвостатом ядре, бледном шаре, мозжечке и др.) сопровождается дегенеративными изменениями с характерной неврологической симптоматикой. Одновременно с поражением центральной нервной системы (ЦНС) происходит накопление меди в роговице в виде пигментации золотисто-коричневого и зеленого цвета по периферии роговицы (кольцо Кайзера–Флейшера). Увеличение экскреции меди с мочой влечет за собой ее отложение преимущественно в проксимальных отделах почечных канальцев и повреждение их эпителия с развитием проксимальной канальцевой дисфункции – синдрома Фанкони.
КЛАССИФИКАЦИЯ БОЛЕЗНИ ВИЛЬСОНА–КОНОВАЛОВА
Особенности клинического течения БВК, проявляющиеся выраженным полиморфизмом, затрудняют создание единой ее классификации. В зависимости от вовлечения в патологический процесс печени, ЦНС и характера экстрапирамидной симптоматики, выделяют пять форм гепатоцеребральной дистрофии: абдоминальную, ригидно-аритмо-гиперкинетическую (раннюю), дрожательно-ригидную, дрожательную и экстрапирамидно-корковую [1, 4]. Согласно Европейской ассоциации по изучению болезней печени (EASL), различают бессимптомную, абдоминальную, церебральную и смешанную формы заболевания [22].
Выделяют 3 генотипических типа БВК. Славянский тип начинается в 20–35 лет и характеризуется неврологическими проявлениями и незначительными поражениями печени. Западный тип БВК манифестирует в 10–16 лет с первичного поражения печени и затем проявляется неврологическими симптомами. Наконец, атипичная БВК сопровождается в основном умеренным снижением уровня церулоплазмина без клинических признаков заболевания [23].
В течении БВК можно выделить латентную стадию, продолжительностью в среднем 5–7 лет, стадию клинических (печеночных, неврологических, почечных и др.) проявлений и терминальную стадию [4]. При эффективном лечении дополнительно выделяют стадию отрицательного баланса меди. В курабельных случаях наблюдается регресс клинических и лабораторных проявлений заболевания [1].
БВК может протекать остро и хронически. Для хронического течения свойственно медленное развитие и прогрессирование заболевания.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Наиболее частой формой (в 50–80% случаев) манифестации БВК является абдоминальная, она встречается в возрасте 5–18 лет. Поражение печени при БВК часто проявляется симптомами хронического прогрессирующего гепатита, реже встречаются асимптомное течение с небольшими биохимическими отклонениями, острый и хронический гепатит с исходом в цирроз печени, фульминантная печеночная несостоятельность [1, 22, 24, 25]. Последний вариант абдоминальной формы чаще наблюдается у женщин, чем у мужчин (4:1), и характеризуется молниеносным течением с летальным исходом, что требует срочной трансплантации печени [26].
Церебральная форма БВК отмечается в 30–50% случаев, обычно на втором–третьем десятилетии жизни. Возможно как моносимптомное ее начало (с тремора головы, конечностей, атаксии и дистонии), так и развитие широкого спектра неврологических, поведенческих или психических расстройств в сочетании с абдоминальной симптоматикой [1, 4, 27].
В 10% случаев встречается почечная симптоматика БВК, которой сопутствуют канальцевые нарушения, снижение клубочковой фильтрации, нефролитиаз, периферические отеки, микрогематурия, незначительная протеинурия, повышение концентрации креатинина крови, аминоацидурия, почечная глюкозурия [28].
Гематологические проявления БВК характеризуются гемолизом, анемией, тромбоцитопенией, коагулопатиями. Кумбс-негативная гемолитическая анемия может быть единственным начальным симптомом заболевания. У 15% больных БВК может развиться острый внутрисосудистый гемолиз [29].
Поражение эндокринной системы при БВК сопровождается аменореей или дисменореей, спонтанными абортами, задержкой полового развития, гинекомастией, гирсутизмом, ожирением, акромегалией, гипопаратиреоидизмом, поражение сердечно-сосудистой системы – кардиомиопатиями, аритмиями, мышечно-скелетной – остеопорозом и артропатиями, желудочно-кишечной – холелитиазом, панкреатитом, спонтанным бактериальным перитонитом. К дерматологическим проявлениям БВК относятся симптом «голубых лунок» у ногтевого ложа, гиперпигментация кожи, сосудистая пурпура, acantosis nigricans, офтальмологическая симптоматика включает роговичное кольцо Кайзера–Флейшера и катаракту, вызванную отложением меди в капсуле хрусталика [1, 4, 24, 30, 31].
ДИАГНОСТИКА
БВК необходимо включать в диагностический поиск при следующих заболеваниях и состояниях: гепатите и циррозе печени неуточненной этиологии; фульминантной печеночной недостаточности; необъяснимом повышении уровня печеночных аминотрансфераз; наличии неустановленной этиологии неврологических и/или психических расстройств с признаками поражения печени; артритах крупных суставов неясного генеза; неуточненных эндокринных нарушениях; необъяснимой приобретенной гемолитической анемии, особенно при отрицательной пробе Кумбса; отягощенном семейном анамнезе по БВК. Возраст пациента при этом не является критерием исключения БВК.
Диагностика заболевания включает клиническое и биохимические исследования. Большое значение имеют клинические проявления поражения печени, нервной системы, остеопороз и кольца Кайзера–Флейшера на роговице (при осмотре в щелевой лампе). Последние выявляются у 90% пациентов с неврологическими симптомами и у 40–50% пациентов с печеночной манифестацией заболевания, они характерны для поздних стадий БВК и могут определяться при хронических холестатических заболеваниях печени [1, 4].
Важное диагностическую роль играют и данные семейного анамнеза, а именно наличие аналогичного заболевания у кровных родственников первой степени родства.
Изменения в стандартных лабораторно-биохимических тестах при БВК неспецифичны. Общеклинический анализ крови с подсчетом тромбоцитов и проба Кумбса позволяют оценить гематологические нарушения, определить наличие Кумбс-негативной гемолитической анемии. Биохимический анализ крови с оценкой показателей цитолиза (аланиновая и аспарагиновая трансаминазы, билирубин), холестаза (щелочная фосфатаза, γ–глутамилтранспептидаза), белковых фракций, коагулограммы позволяет выявить степень активности воспалительного процесса в паренхиме печени и печеночно-клеточной недостаточности.
Наличие отрицательных результатов анализа крови на маркеры вирусных гепатитов, аутоиммунных заболеваний печени, исключение алкогольного или медикаментозного поражения печени, гемохроматоза (особенно у молодых лиц) должны инициировать проведение обследования на БВК.
Специфическими для БВК являются показатели обмена меди: снижение церулоплазмина сыворотки (<0,1 г/л) и общей меди (<0,8 мг/л), повышение уровня свободной меди сыворотки (>0,15 мг/л, за исключением фульминантной печеночной недостаточности), возрастание суточной экскреции меди с мочой (>0,64 мкмоль/сут при латентной стадии и >1,6 мкмоль/сут при наличии клинических проявлений), повышение содержания меди в ткани печени (более 4 мкмоль/г сухой массы), возрастание меди ликвора (>1 мкмоль/л при церебральной форме). Для диагностики и мониторинга терапии БВК может использоваться проба с D-пеницилламином. Диагностическим считается повышение суточной экскреции меди с мочой: базальной – более 50 мкг/сут; в пробе с D-пеницилламином (500 мг 2 раза/сут) – более 1600 мкг/сут [1, 4].
Дополнительными методами диагностики БВК служат:
- ультразвуковое исследование органов брюшной полости (оценка размеров и структуры печени, селезенки, состояния сосудов портальной системы, определение наличия асцитической жидкости, лимфаденопатии);
- эзофагогастродуоденоскопия (выявление признаков портальной гипертензии и наличия сопутствующей гастродуоденальной патологии);
- ультразвуковая допплерография сосудов брюшной полости (изучение состояния портального кровотока);
- рентгенологическое исследование органов грудной клетки;
- фиброэластометрия печени (оценка степени фиброза печени);
- компьютерная томография головного мозга (расширение желудочков, атрофия коры и ствола мозга, а также билатеральные зоны пониженной плотности в области базальных ганглиев);
- магнитно-резонансная томография головного мозга (характерные очаги в головном мозге на фоне некоторой атрофии коры);
- магнитно-резонансная спектроскопия (накопление меди и снижение индексов N–ацетиласпартат/креатин и холин/креатин в бледных шарах);
- позитронно-эмиссионная томография (снижение активности допа-декарбоксилазы);
- биопсия печени (определение содержания меди);
- сцинтиграфическое исследование биоптатов печени с радиоактивной медью – тест включения изотопа меди (64Cu или 67Cu) в церулоплазмин (позволяет выявлять больных БВК и гетерозиготных носителей).
Показания для консультации специалистов:
- консультация офтальмолога и осмотр с помощью щелевой лампы (для выявления колец Кайзера–Флейшера);
- консультация невролога (оценка неврологического статуса);
- консультация психотерапевта (оценка и коррекция психологических проблем);
- консультация психиатра (оценка и коррекция психических расстройств);
- консультация гастроэнтеролога (коррекция нарушений желудочно-кишечного тракта);
- консультация гематолога (при наличии гемолитической анемии, коагулопатии);
- консультация нефролога (при патологии в анализах мочи);
- консультация хирурга (при риске пищеводно-желудочных кровотечений, для определения показаний к трансплантации печени);
- консультация инфекциониста (при сопутствующем вирусном гепатите);
- консультация гинеколога (при наличии акушерско-гинекологических патологий).
Существенная роль в подтверждении диагноза БВК отводится молекулярно-генетическому исследованию. Рекомендуется начинать диагностический поиск патогенных мутаций гена ATP7B с исследования четырех наиболее частых мутаций: c.3207C>A, c.3190G>A, c.3402delC и c.2304insC [32].
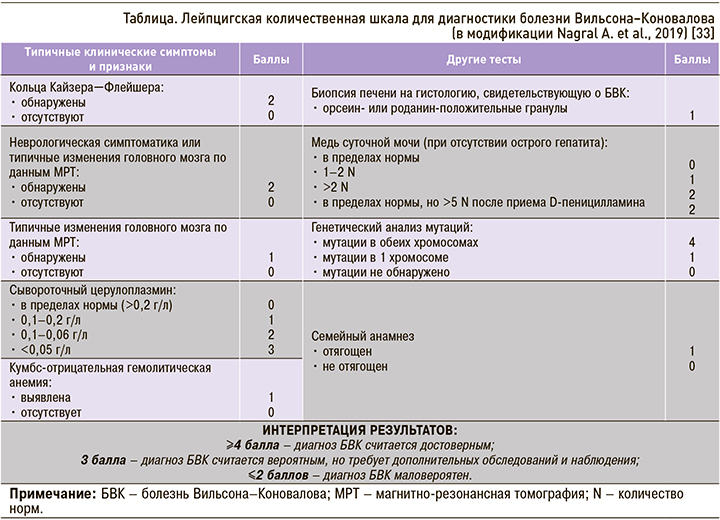
Для постановки диагноза БВК требуется учитывать совокупность клинических проявлений болезни, результаты лабораторных, инструментальных методов исследования и данные молекулярно-генетического анализа. С целью стандартизации диагностики заболевания в 2001 г. была предложена Лейпцигская балльная шкала, согласно которой диагноз БВК устанавливается путем оценки выявленных показателей. В 2019 г. Nagral A. et al. модифицировали эту диагностическую шкалу (табл.): в нее были добавлены баллы за семейный анализ, заставляющий заподозрить БВК, исключена оценка меди в печени, скорректированы баллы за показатели церулоплазмина в сыворотке крови [33].
ПРИНЦИПЫ ЛЕЧЕНИЯ
Лечение БВК должно быть своевременным, непрерывным и пожизненным.
Важным компонентом терапии служит диета, ограничивающая поступление меди в организм, особенно в первый год лечения. Из пищевого рациона исключаются продукты с высоким содержанием этого микроэлемента (свыше 0,5 мг/100 г), используется деионизированная вода (если содержание меди превышает 0,2 ppm). Не рекомендуется пользоваться медной посудой для хранения воды и пищевых продуктов. Нежелательно и применение медьсодержащих внутриматочных устройств, препаратов, содержащих эстрогены, поливитаминных комплексов, включающих микроэлементы, пищевых добавок и средств искусственного питания [1, 4, 22].
Современная медикаментозная терапия БВК подразделяется на начальную и поддерживающую. На данный момент в мировой практике используются медь-элиминирующие препараты: хелатирующие препараты общего действия, вызывающие купрурию (D-пеницилламин, триентин), индукторы металлотионеина, блокирующие кишечное всасывание меди (препараты цинка), хелатор двойного действия тетратиомолибдат и унитиол [1, 22, 24].
При наличии клинических симптомов лечение начинают с хелатирующих агентов (D-пеницилламина или триентина) [1, 10, 25]. D-пеницилламин предпочтительнее принимать натощак (за 1 ч до либо через 2 ч после еды, поскольку прием пищи снижает его абсорбцию) в индивидуально подобранной дозе – в пределах от 1,5 до 4 г/сут. Оптимальной считается дозировка 0,9–1,2 г/сут. Адекватность лечения должна мониторироваться измерением суточной экскреции меди с мочой. Учитывая антипиридоксиновый эффект D-пеницилламина, к терапии необходимо добавлять пиридоксин (витамин В6) в дозе 25–50 мг/сут внутрь.
У трети пациентов терапия D-пеницилламином сопряжена с целым рядом побочных эффектов, и в этом случае возможно назначение триентина [10, 22, 24]. Однако данный препарат в настоящее время в России не зарегистрирован.
При бессимптомной стадии болезни на этапе поддерживающей терапии (как в виде монотерапии, так и в комбинации с D-пеницилламином), а также при непереносимости и толерантности к хелатирующим средствам целесообразно назначать препараты цинка [1, 34]. Преимущества их заключаются в меньшей токсичности и низком риске развития побочных эффектов, а переносимость зависит от используемой соли цинка (сульфат, ацетат или глюконат). Рекомендуемая доза составляет 150 мг в пересчете на элементарный цинк в сутки в 3 приема за 30 мин до еды.
Тетратиомолибдат аммония (ТТМ) – мощный хелатор, к сожалению, предыдущие клинические испытания препарата не увенчались успехом [25, 35]. Новым терапевтическим вариантом является тетратиомолибдат бисхолина, который в настоящее время активно изучается. Его механизм действия включает образование комплексов стабильная медь–ТТМ–альбумин, ингибирование поглощения меди печенью и нейронами, а также повышение экскреции меди с желчью [36].
В качестве вспомогательной терапии БВК возможно назначение витаминов В1 и В6 (избыточные количества меди блокируют их активность), антиоксидантов (витамин Е), гепатопротекторов и унитиола (димеркаптопропансульфоната натрия) [4]. Последний за счет наличия двух сульфгидрильных групп способен восстанавливать функции ферментных систем печени, а также обладает детоксицирующим эффектом.
Трансплантация печени показана при неэффективности терапии БВК в течение нескольких месяцев у пациентов с декомпенсированным циррозом, развитии прогрессирующей недостаточности печени, фульминантной печеночной несостоятельности, прогрессирующих и необратимых неврологических нарушениях. Выживаемость после оперативного лечения составляет 79–87% в течение первого года, пятилетняя выживаемость сопоставима с годовой [1, 28].
ЗАКЛЮЧЕНИЕ
Таким образом, БВК – редкий пример наследственного заболевания, для которого разработаны высокоэффективные методы лечения. Систематическое применение этих методов позволяет достичь уменьшения всех симптомов, вплоть до регресса, а следовательно, перевести заболевание в разряд курабельных. Повышение осведомленности практикующих врачей о БВК, их ориентация на выявление ранних признаков заболевания и строгий контроль лечения являются жизненно важными стратегиями для улучшения прогноза данных пациентов.


