
Известно, что тенасцин-Х – гликопротеин семейства тенасцинов, который выполняет главную архитектурную функцию между волокнистыми структурами экстрацеллюлярного матрикса (ЭЦМ) с элементами клеточной адгезии, миграции, роста и дифференцировки через модуляцию сигнальных путей (например, через изменение биодоступности трансформирующего фактора роста – TGF-β) [1].
Выделяют 4 типа тенасцинов:
- тенасцин-R специфичен для центральной нервной системы;
- тенасцин-C является «онкофетальным» белком, контролируемым факторами роста, цитокинами, механическим стрессом и т.д., но с ограниченным появлением в пространстве и времени;
- тенасцин-Х служит составным компонентом соединительной ткани, и его уровень практически не зависит от внешних факторов;
- тенасцин-W подобен тенасцину-C, но его экспрессия еще более ограничена [2].
Основная функция тенасцина-Х – взаимодействие с волокнами экстрацеллюлярного матрикса (ЭЦМ), такими как коллаген и эластин, что повышает прочность тканей. Например, у нокаутированных по тенацину-Х мышей были обнаружены снижение прочности влагалищной стенки и кожи [3].
Тенасцин-Х кодируется геном TNXB4, расположенном на хромосоме 6p21 [1]. Полный дефицит TNX приводит к развитию рецессивной формы синдрома Элерса–Данлоса, а у гетерозигот – к гипермобильному фенотипу и пузырно-уретральному рефлюксу [3 ,4]. Долгое время считалось, что гипермобильный тип синдрома Элерса–Данлоса (EDS-HT) и синдром семейной доброкачественной гипермобильности суставов – два различных синдрома. Однако дальнейшие исследования показали сильную корреляционную взаимосвязь между клиническими проявлениями при этих синдромах от уровня тенасцина-X в сыворотке крови, что является результатом гаплонедостаточности или дефицита гена TNXB [5].
EDS-HT считался наименее тяжелым проявлением классического типа синдрома Элерса–Данлоса. Из осложнений EDS-HT следует выделить подвывихи и вывихи суставов, которые возникают спонтанно или с минимальной травмой, а также гастропарез, грыжу пищеварительного тракта, ортостатическую гипотензию, позиционную ортостатическую тахикардию и ортостатическую непереносимость, сосудистые нарушения и т.д. [6, 7, 8].
Примечательно, что все эти клинические проявления EDS-HT объединяет слабость или гипотония тканей. Это обусловлено дефицитом тенасцина-Х, который локализуется в соединительных тканях: перитендинуме, эпимизии, перимизии, дерме кожи, слизистой оболочке мышц, клубочках почек и вокруг кровеносных сосудов, что и объясняет клинические проявления гипермобильных фенотипов таких синдромов, как синдром гипермобильности суставов и EDS-HT [1].
В литературе клинические проявления, описанные в рамках дефицита тенасцина-Х, частично укладываются в Вилльфраншские критерии диагностики: кожные критерии (гиперэластоз, истончение, грыжи, пролапсы), сердечно-сосудистые критерии, а также гипермобильность суставов [9].
Несмотря на четкость проявления элерсоподобных критериев и других сопутствующих выраженных клинических проявлений недифференцированной дисплазии соединительной ткани (ДСТ), фенотипически отнести больных к какой-либо не только моногенной форме синдрома Элерса–Данлоса, но и тем более к какому-либо фенотипу часто не представляется возможным. Поэтому поиск новых методов диагностики для подтверждения формы ДСТ крайне актуален.
Нами были обследованы 2 пациентки с элерсоподобным фенотипом и пролапсом гениталий, поступившие в гинекологическую клинику для оперативного лечения. Панель полиморфизмов и генетических мутаций была создана на основе метода высокопроизводительного секвенирования (NGS). Благодаря ей возможен анализ по следующим точкам: B4GALT7, BMP1, C1R, COL1A1, COL1A2, COL11A1, COL11A2, COL3A1, COL5A1, COL5A2, ELN, FBLN4, FBLN5,FBN1, FBN2, FGFR3, FLNA, MYH11, PLOD1, TGFB1, TGFBR1, TGFBR2, TNXB+ ACTA2, ADAMTS10, ADAMTS2, ADAMTSL4, COL2A1, DCHS1, LOX, MMVP1, MMVP3, MYLK, SMAD3, COL1A1 rs1800012, TGFBrs1800471, COL3A1 rs1800255, FBLN5 rs2018736, FBLN5 rs12589592, COL2A1 rs2276455, COL2A1 rs63118460 (rs7963636), MMP10 rs17435959, MMP10 rs17293607, ESR1 rs2228480, PGRrs484389, MMP13 rs2252070, GDF5 rs143383, MMP3 rs35068180, MMP3 rs3025058, VEGFArs699947, VEGFA rs2010963, VEGFA rs3025039, MMP9 rs17576, MMP9 rs3918242, LAMC1 rs10911193.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ №1
Больная П., 62 лет, поступила в клинику для оперативного лечения пролапса гениталий.
Клинический диагноз: неполное выпадение матки и стенок влагалища с началом формирования энтероцеле, ректоцеле 2–3 степени, цистоцеле 3–4, синдром протрузии и релаксации тазового дна.
Из анамнеза: росла и развивалась нормально. Менструация установилась с 12 лет, без особенностей. Беременность одна, роды одни.
Роды в анамнезе (1983) были своевременные, самопроизвольные, в затылочном предлежании, вес/рост ребенка при рождении составляли 3200 г/53 см. Роды протекали с развитием слабости родовой деятельности, что потребовало родостимуляции окситоцином. Длительность родов – 12 ч, промежность цела. Через 10 лет после родов пациентка отметила опущение стенок влагалища. Заболевание прогрессировало в течение 10 лет.
Перенесенные заболевания: артериальная гипотензия. Рабочее артериальное давление (АД) 90/60 мм рт.ст., пациентка имеет склонность к простудным заболевания, страдает хроническим гастритом, хроническим тонзиллитом, ретикулярной болезнью вен нижних конечностей
Наследственность: у матери было недержание мочи.
Результаты физикального обследования: нормальное сложение, пониженное питание, рост 168 см, вес 64 кг. Кожа тонкая, эластичность обычная, сосудистая сеть не выражена, наблюдаются единичные папиросные рубцы, стрий нет.
Осанка ослаблена. Больная отмечает непереносимость статических нагрузок, предпочитая динамические, имеет феномен «плоской спины».
Арахнодактилия и долихостеномелия не выявлены: размах рук 170 см (менее 7 см), длина среднего пальца кисти 9 см, длина нижнего сегмента тела 88 см, тест большого пальца отрицательный, симптом запястья положительный для всех пальцев.
Гипермобильность суставов 3–4 балла (62 года), вывихи в анамнезе пациентка отрицает, отмечает артралгии в крупных суставах без воспалительных проявлений.
Имеется готическое нёбо, выраженный кариес.
Результаты инструментального исследования:
- электрокардиография (ЭКГ) – ритм синусовый;
- эхокардиография (ЭхоКГ) – пролапс митрального клапана (ПМК) 2 степени, пролапс трикуспидального клапана (ПТК) 2 степени, митральная регургитация (МР) 2 степени, трикуспидальная регургитация (ТР) 2 степени, регургитация на клапане легочной артерии 1 степени, признаки фиброза створок митрального клапана. Фракция выброса (ФВ) 56%, корень аорты 37 мм.
Учитывая пролапс гениталий на фоне синдрома протрузии и релаксации тазового дна, выполнены дополнительные специальные исследования.
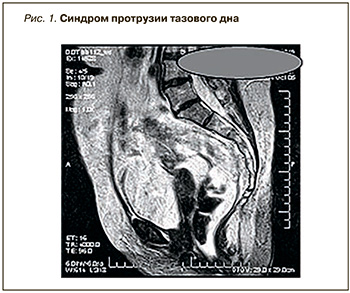 Данные дефекографии в положении стоя: прямая кишка опускается вниз до 7,8 см, складки слизистой собираются на уровне верхней 1/3 анального канала, формируя «воронку». Диагноз: R-логические признаки опущения промежности, внутренней ректальной инвагинации (рис. 1).
Данные дефекографии в положении стоя: прямая кишка опускается вниз до 7,8 см, складки слизистой собираются на уровне верхней 1/3 анального канала, формируя «воронку». Диагноз: R-логические признаки опущения промежности, внутренней ректальной инвагинации (рис. 1).
При сфинктерометрии отмечено снижение тонуса анального сфинктера, выявлены признаки недостаточности анального сфинктера. При нейрофизиологическом исследовании выявлены признаки нейропатии пудендального нерва.
При генетическом анализе выявлен миссенс вариант в гене TNXB.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ №2
Больная А., 58 лет, поступила в клинику для оперативного лечения пролапса гениталий с диагнозом «опущение стенок влагалища: цистоцеле 2–3, ректоцеле 3 степени. Недостаточность анального сфинктера 1 ст. Недержание газов. Проктогенные запоры, долихосигма».
Из анамнеза: росла и развивалась нормально. Менструация установилась с 13 лет (без особенностей). Беременностей – 2, родов – 2.
Первые роды (1982) были своевременные, самопроизвольные, в затылочном предлежании. Вес/рост новорожденного составляли 3900 г/55 см. Имела место эпизиотомия, заживление первичным натяжением.
Вторые роды (1985) своевременные, самопроизвольные, в затылочном предлежании. Вес/рост новорожденного – 4100 г/56 см.
Через 22 года после вторых родов был выявлен пролапс гениталий.
Сопутствующие заболевания: нормотоник, рабочее АД 110/70 мм рт.ст. (максимально не более 130/90 мм рт.ст.), имеется склонность к простудным заболевания (2–3 раза в год), тонзиллэктомия в анамнезе в возрасте 16 лет, миопия 3,5 д в сочетании с астигматизмом с 14 лет, долихосигма, сигмоидоцеле (выпадение сигмовидной кишки в дугласов карман).
Результаты физикального обследования: нормальное сложение, пониженное питание. Рост 156, вес 56 кг. Кожа тонкая, суховата, отмечается склонность к легкому травмированию, предрасположенность к легкому образованию «синяков», сосудистая сеть не выражена, стрий нет.
Осанка сохранена, феномен «плоской спины» отсутствует.
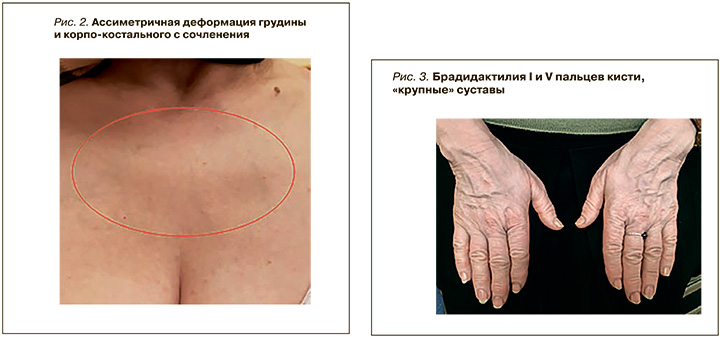
Наблюдаются малые аномалии развития: деформация грудины – ассиметрия корпокостальная (рис. 2), брахидактилия V пальцев кистей рук (рис. 3).
Арахнодактилия и долихостеномелия не выявлены: размах рук 155 см (менее 7 см), длина среднего пальца кисти 8 см, длина нижнего сегмента тела 80 см, тест большого пальца отрицательный, симптом запястья положительный для всех пальцев.
Гипермобильность суставов – 0 баллов (58 лет), вывихи в анамнезе отрицает, страдает артрозоартритом 2 степени суставов кистей рук (см. рис. 3), остеохондрозом шейного отдела позвоночника с 20 лет.
Кариес не выражен.
Результаты инструментального исследования:
- ЭКГ – ритм синусовый;
- ЭхоКГ – фиброз створок аортального клапана, уплотнение створок митрального клапана, ПМК 1 степени, ФВ 68%, другие клапаны не изменены, в полости левого желудочка – дополнительная хорда левого желудочка.
Учитывая наличие пролапса гениталий, были выполнены дополнительные специальные исследования. При дефекографии выявлены R-логические признаки опущения промежности в стадии компенсации, долихосигма. При нейрофизиологическом исследовании признаки нейропатии пудендальных нервов не обнаружены.
При генетическом анализе выявлен миссенс-вариант в гене TNXB, мутация в гене Col 11A2.
ОБСУЖДЕНИЕ КЛИНИЧЕСКИХ СЛУЧАЕВ
Обе пациентки имели пролапс гениталий и мутацию в гене TNXB. Это не противоречит литературным данным, так как у больных с дефицитом тенасцина-Х в 86% случаев развивается пролапс гениталий, а в 40% в анамнезе отмечаются послеродовые кровотечения [3, 10, 11]. Слабость стенок влагалища авторы наблюдали и у нокаутированных по тенасцину-Х мышей [3]. Аналогичные закономерности отмечены и для больных с синдромом Элерса–Данлоса [12, 13].
Как было сказано выше, EDS-HT и доброкачественная семейная гипермобильность суставов имеют общую сниженную экспрессию тенасцина-Х, а также отсутствие генетических локусов, характерных для других форм синдрома Элерса–Данлоса [5]. Учитывая, что у обеих больных выявлена мутация в гене TNXB, вероятность того, что они имели элерсоподобный фенотип, крайне высока.
Пациентка П., 62 лет (клинический случай № 1), в наибольшей степени соответствовала элерсоподобному фенотипу: у нее наблюдались выраженные кожные проявления, мышечная гипотония и гипотрофия, артериальная гипотензия, синдром гипермобильности суставов 4 балла в 62 года, синдром протрузии и релаксации мышц тазового дна с развитием апикальной формы пролапса гениталий, клапанный синдром с наличием регургитации 2 степени на двух и более клапанах, а также наследственный характер пролапса гениталий при единственных не осложненных родах некрупным (3200 г) плодом. Признаки марфаноподоного фентипа (марфаноидной внешности) у больной отсутствовали. В клинические проявления элерсоподобного фенотипа, дефицита тенасцина Х и пролапса гениталий не укладывалась лишь пудендальная нейропатия, которая могла быть самостоятельной причиной пролапса гениталий и являться следствием травматичных родов.
Пациентка А., 58 лет (клинический случай № 2), также имела пролапс гениталий, однако локального типа (цисторектоцеле без апикального маточного пролапса), который не характерен для элерсоподоного фенотипа [14]. Локальная форма пролапса гениталий могла быть следствием родовой травмы (крупные новорожденные – 3900 г и 4100 г). Учитывая, что рост пациентки составляет 156 см, можно однозначно утверждать, что она имела общеравномерносуженный таз, а потому роль травмы в родах в генезе пролапса гениталий не могла быть изначально отвергнута.
Тем не менее при нейрофизиологическом исследовании у данной пациентки пудендальная нейропатия обнаружена не была, вследствие чего родовая травма была исключена как возможная первопричина локальной формы пролапса гениталий.
Кроме того, у пациентки имелось клинически значимое ректоцеле 3 степени, и специальные дополнительные методы обследования показали, что это сигмоидоцеле (выпадение в дугласов карман сигмовидной кишки). Наличие сигмоидоцеле указывало на скрытую апикальную форму пролапса гениталий, которая, как правило, развивается при элерсоподобном фенотипе.
Также эта больная предъявляла жалобы на проктогенные запоры и одновременно анальное недержание, что указывало на желудочно-кишечную дисфункцию преимущественно моторного типа, характерную для элерсоподобных проявлений [15].
По мнению авторов, именно снижение экспрессии тенасцина-Х, который находится в ассоциации с холинергическими кишечными нейронами, осуществляющими моторный контроль, служит причиной нарушений моторного типа. В эксперименте было показано, что мыши с дефицитом TNX имели внутренний пролапс прямой кишки на фоне потери сократительной способности дистального отдела толстой кишки. Авторы сделали вывод, что экстрацеллюлярный матрикс – не просто опорная структура, но и неотъемлемая часть микроокружения для мотонейронов толстой кишки.
На элерсоподобный фенотип у больной А., 58 лет, также указывали миопия и астигматизм, манифестировавшие в пубертатном возрасте, склонность к простудным заболеваниям с тонзиллэктомией в анамнезе, патология клапанного аппарата сердца с наличием регургитации на клапанах.
Как и больная П., пациентка А. не имела признаков марфаноподобного фенотипа (внешности), однако у нее наблюдались малые аномалии развития (дополнительную хорду в левом желудочке, деформацию грудины, брахидактилию, долихосигму, рост ниже среднего).
У обеих пациенток был выявлен синдром протрузии и релаксации тазового дна по данным дефекографии. Но если у первой больной это могло быть следствием диагностированной пудендальной нейропатии (по результатам нейрофизиологического исследования), то у второй нарушения проводимости по тазовым нервам выявлено не было, что представляет особый клинический интерес.
Если вторая больная (А.) родила двух крупных детей (3900 г и 4100 г), перенеся эпизиотомию, и при этом и не имела пудендальной нейропатии, то вопрос о возникновении у первой пациентки (П.) пудендальной нейропатии после единственных неосложненных родов без эпизиотомии (вес ребенка 3200 г) также вызывал научный интерес.
Выше было показано, что дефицит тенасцина-Х непосредственно изменяет моторику кишечника в сторону гипокинеза. Этот механизм опосредован через холинергическую иннервацию.
В 2009 г. Voermans N.C., анализируя причину нервно-мышечных расстройств у больных с синдромом Элерса–Данлоса (n=40), показал, что в 13% случаев при исследовании нервной проводимости в основе мышечной слабости, гипотонии и повышенной мышечной утомляемости лежала аксонная полиневропатия. При игольчатой электромиографии в 60% случаев были выявлены смешанные нейрогенно-миопатические нарушения. При УЗИ мышц в 50% случаев была обнаружена атрофия мышц, а при биопсии в 28% случаев – аномалии в мышечном или нервном внеклеточном матриксе (миопатии) [16]. Это еще раз подтверждает вывод Rubina A. (2018) о том, что ЭЦМ служат не просто опорными структурами, но и неотъемлемой частью микроокружения для мотонейронов. Поэтому синдром протрузии и релаксации тазового дна у обеих больных мог быть обусловлен нейрогенно-миопатическими нарушениями из-за нарушений микроокружения в ЭЦМ вследствие дефицита тенасцина-Х. В 2001 г. нами было показано, чем выраженней были клинические проявления ДСТ у пациенток с пролапсом гениталий, тем чаще встречался нейрогенный мочевой пузырь, т.е. недержание мочи имело смешанный нейрогенно-мышечный характер [17].
Другой, не менее актуальный и интересный вопрос, возникший в наших наблюдениях, – отсутствие гипермобильности суставов у пациентки А. (клинический случай № 1) при дефиците тенасцина-Х, затруднявшее постановку диагноза «элерсоподобный фенотип», несмотря на протрузию тазового дна и спланхноптоз. У этой больной, помимо миссенс-мутации в гене TNXB, была также найдена мутация в гене Col 11A2, расположенном на 6p21.32 хромосоме и кодирующем синтез коллагена XI типа. Коллаген XI типа выступает компонентом хряща, который обнаруживается в суставах и позвоночнике, внутреннем ухе и т.д. [18]. Аутосомно-доминантная форма приводит к развитию отоспондиломегаэпифизарной дисплазии (например, синдрома Стиклера и т.д.), а аутосомно-рецессивная – к развитию дегенеративных заболеваний суставов, напоминающих остеоартрит, который проявляется в раннем взрослом возрасте и поражает преимущественно крупные суставы: бедренные, коленные, локтевые, плечевые [18]. Напомним, что больная А. имела остеоартроз суставов кисти (см. рис. 3), а также остеохондроз, который у нее манифестировал в возрасте 20 лет. Вполне возможно, что эта мутация, а также аутоиммунные процессы, происходящие при этой мутации, могли повлиять на связочный аппарат суставов и частично нивелировать их гипермобильность.
Vikkula et al. на примере 3 сибсов продемонстрировал, что замена глицила на аргинин у гомозигот приводила к формированию специфичного фенотипа: короткие конечности, рост взрослого ниже среднего, увеличение поясничного лордоза, формирование крупных межфаланговых суставов, короткие пястные кости и т.д. [19]. За малым исключением, похожие клинические проявления, такие как малый рост, брахидактилия мизинцев и больших пальцев кисти, остеоартроз кистевых суставов, были представлены у второй больной (см. рис. 3).
ЗАКЛЮЧЕНИЕ
Таким образом, в данной работе показана клиническая взаимосвязь нескольких генетических мутаций. Мутации в гене COL11A2 могут нивелировать выраженность синдрома гипермобильности суставов, но формируют другие признаки в зависимости от локализации и уровня мутации. В данном случае это были остеоартроз, ранний остеохондроз, артрозоартрит, которые являлись специфичными при рецессивной мутации данного гена.
Пудендальная нейропатия, так же как и проктогенные запоры и другие моторные нарушения толстой кишки, у пациенток с пролапсом гениталий могут быть не только следствием ректоцеле, а результатом дефицита экспрессии тенасцина-Х.
Диагностика ДСТ, которая относится к полисимптомным мультиорганным заболеваниям, имеющим нередко стертые перекрестные клинические проявления, нуждается в поиске новых методов.
Разработанная панель полиморфизмов и генетических мутаций для постановки диагноза ДСТ и идентификации ее формы позволяет не только установить генетический дефект, но и объяснить перекрестные клинические проявления и их взаимодействия в результате нескольких мутаций.


