
Современная распространенность метаболических заболеваний во всем мире приобретает масштабы пандемии. Рассматривая различные метаболические нарушения, ассоциированные с избыточным весом или накоплением жировой ткани в воротах печени и на сальнике (т.е. с висцеральным ожирением) в комплексе с прогрессирующим состоянием инсулинорезистентности, исследователи используют термин метаболический синдром (МС) [1].
Основные заболевания и состояния, входящие в МС: ожирение, сахарный диабет 2 типа (СД 2), дислипидемия и артериальная гипертензия (АГ). Учитывая, что объединяющей патофизиологической основой этих нарушений выступают инсулинорезистентность (ИР), компенсаторная гиперинсулинемия и гипоадипонектинемия, становится более очевидной их связь с ранним развитием сердечно-сосудистых заболеваний (ССЗ) [1]. Одной из наиболее часто развивающихся кардиоваскулярных патологий у пациентов с МС является сердечная недостаточность [2].
Связь метаболических нарушений с развитием сердечной недостаточности была показана еще более четырех десятилетий назад, когда у пациентов с СД 2, не страдавших АГ или ишемической болезнью сердца (ИБС), были выявлены гипертрофия миокарда левого желудочка (ГЛЖ) и/или сократительная дисфункция [3]. Морфологическим субстратом таких нарушений служит развитие и прогрессирование метаболической кардиомиопатии, характерной для СД 2. Благодаря совершенствованию диагностических возможностей, на современном этапе клиницист может диагностировать диабетическую кардиомиопатию у пациента с СД 2 независимо от наличия АГ и ИБС [4].
Механизм развития этого вида кардимиопатии заключается в избыточном накоплении липидов и их дальнейшем токсическом воздействии на миокард. Данное состояние получило название кардиальный стеатоз; скорость его развития отражает степень несоответствия между уровнями поступления и утилизации жирных кислот в миокардиоцитах. Таким образом, учитывая, что миокардиоциты не специализируются на длительном хранении липидов, а чрезмерное поглощение жирных кислот приводит к быстрому увеличению количества внутриклеточных липотоксичных метаболитов, по сути происходящих процессов развивающуюся кардиомиопатию можно отнести к липотоксическому виду [5]. Такой же механизм поражения миокарда может наблюдаться и у больных с инсулинорезистентностью, ожирением, дислипидемией, но без СД 2. Однако ставить диагноз диабетической кардиомиопатии таким пациентам нельзя ввиду отсутствия диабета, хотя патогенетически в миокарде будут протекать процессы, связанные с кардиальным стеатозом и липотоксичностью [5, 6]. Необходимо также подчеркнуть, что в отличие от широко изучаемых сегодня атеросклеротических ССЗ отличительной чертой липотоксической кардиомиопатии (ЛК) является именно накопление токсических липидных метаболитов в миокардиоцитах [7].
Основные клинические проявления на начальных этапах развития ЛК включают постепенное развитие ГЛЖ и диастолической дисфункции миокарда, которая в дальнейшем переходит в сердечную недостаточность с сохраненной фракцией выброса (СНсФВ), а систолическая дисфункция присоединяется на более поздней стадии [7]. Важно отметить, что в реальной клинической практике у большинства пациентов с СНсФВ присутствует хотя бы один из компонентов МС (висцеральное ожирение, инсулинорезистентность, дислипидемия или СД 2), что позволяет предполагать наличие взаимосвязи между ЛК и развитием диастолической дисфункции миокарда [5].
Таким образом, заболевания, связанные патогенезом с инсулинорезистентностью, могут приводить к увеличению накопления токсических метаболитов, которые, в свою очередь, способны запускать различные патологические процессы внутри миокардиоцитов и приводить к развитию сердечной недостаточности. Для лучшего понимания происходящих процессов в миокарде следует подробнее остановиться на патогенетических моментах ЛК.
КЛЮЧЕВОЙ МОМЕНТ ПАТОГЕНЕЗА ЛИПОТОКСИЧЕСКОЙ КАРДИОМИОПАТИИ
Известно, что за счет гибкого механизма переключения между субстратами миокард для получения энергии может использовать как жирные кислоты, так и глюкозу. В условиях достаточного поступления кислорода, т.е. в отсутствие ишемии, в здоровом сердце производство энергии идет в основном за счет утилизации свободных жирных кислот [6]. Однако при развитии МС, т.е. появлении инсулинорезистентности, висцерального ожирения, дислипидемии и СД 2, в миокард устремляется слишком большое количество жирных кислот, концентрация которых превышает физиологический порог их использования [8]. На начальных этапах скорость процессов поступления и утилизации жирных кислот в миокардиоцитах у пациентов с МС многократно возрастает, тогда как утилизация глюкозы снижается; это влечет за собой увеличение потребности миокарда в кислороде. В то же время из-за дисбаланса между поглощением и окислением жирных кислот, развивающегося вследствие этой ситуации, начинается патологическое накопление липидов и токсических липидных метаболитов в миокарде – тот самый кардиальный стеатоз, выступающий главной отличительной чертой патогенеза ЛК и предшествующий возникновению сердечной недостаточности [9].
Основные токсические липидные метаболиты, запускающие развитие повреждений в миокардиоцитах, – это длинноцепочечные ацилкарнитины (ДцАЦ), церамиды (ЦМ) и диацилглицериды (ДАГ). Главные повреждающие эффекты ЦМ и ДАГ связаны с увеличением продукции активных форм кислорода (АФК) и развитием окислительного стресса, а также с усилением внутриклеточных процессов инсулинорезистентности и нарушением митохондриального транспорта электронов, активации процессов апоптоза и фиброза, что способствует клинической прогрессии ГЛЖ и сердечной недостаточности [10, 11]. Своим появлением ЦМ и ДАГ обязаны первичному накоплению ДцАЦ; они являются продуктами неокислительных реакций, в которые вступают при чрезмерном накоплении в миокардиоцитах. Сами ацилкарнитины образуются с целью переноса накопившихся активированных жирных кислот внутрь митохондрий для их дальнейшего участия в процессах b-окисления. Нарушения, которые возникают в результате активного поступления и накопления жирных кислот, существенно повышают уровень ДцАЦ, приводя к ухудшению электрофизиологической функции миокарда, изменению проницаемости митохондриальной мембраны, усилению инсулинорезистентности и развитию воспаления [12]. На сегодняшний день известны результаты исследований, которые продемонстрировали значимую ассоциацию повышенных уровней ДцАЦ с неблагоприятными клиническими исходами у пациентов с хронической сердечной недостаточностью [13].
Таким образом, развивающиеся вследствие продолжительного накопления токсичных липидных метаболитов ограниченная окислительная способность митохондрий, окислительный стресс, инсулинорезистентность и реакции воспаления способствуют дальнейшему развитию и прогрессированию кардиального стеатоза (рис. 1).

ДРУГИЕ ФАКТОРЫ ЛИПОТОКСИЧЕСКОЙ КАРДИОМИОПАТИИ: ГЛЮКОЗОТОКСИЧНОСТЬ, ОКИСЛИТЕЛЬНЫЙ СТРЕСС И ВОСПАЛЕНИЕ
У пациентов с гипергликемией (СД 2 или предиабетом) дополнительно к липидным повреждениям миокардиоцитов развивается состояние глюкозотоксичности, сопровождающееся накоплением токсичных метаболитов глюкозы [14]. Эти токсичные метаболиты вносят вклад в образование большого количества конечных продуктов гликирования (КПГ), которые модифицируют белки и нуклеиновые кислоты, тем самым нарушая функции клеток. КПГ также запускают множество биологических сигнальных путей через свои рецепторы, вызывая выработку воспалительных цитокинов и АФК, способствуют развитию ГЛЖ и сердечной недостаточности (см. рис. 1) [15].
При совместном участии липотоксичных и глюкотоксичных промежуточных продуктов обмена резко возрастает продукция АФК, которые блокируют функционирование эндогенной антиоксидантной системы. Окислительный стресс приводит к дисфункции митохондрий, усилению реакций локального воспаления и апоптозу, что способствует прогрессии гипертрофии миокардиоцитов и сократительной дисфункции [16].
Ожирение и инсулинорезистентность сопровождаются развитием вначале слабой, а затем все более усиливающейся (по мере увеличения длительности инсулинорезистентности) системной воспалительной реакцией. Ей сопутствует аномальная продукция провоспалительных цитокинов, играющих важную роль в патогенезе сердечного фиброза, гипертрофии и сократительной дисфункции у пациентов с ЛК.
При дальнейшем развитии воспалительных реакций фиброз становится одной из ключевых патологических особенностей ЛК на поздней стадии. Известно, что инсулинорезистентность, помимо прочего, характеризуется активацией рецепторов врожденного иммунитета, которые, наряду с клетками иммунной системы, локализованы и в инсулинозависимых тканях. Среди них особое значение имеют Toll-подобные рецепторы, особенно TLR2 и TLR4, количество которых в гепатоцитах, адипоцитах и миоцитах (в том числе в миокардиоцитах) существенно больше. Стимуляция TLR-2 и TLR-4 приводит к нарушению внутриклеточной передачи сигналов инсулина, т.е. к усилению процессов инсулинорезистентности. Эта активация лежит в основе хронического воспаления при ожирении и инсулинорезистентности в результате перехода от адаптивного к неадаптивному иммунометаболизму и аберрантных иммунных реакций с усилением выработки провоспалительных цитокинов [17].
Жирные кислоты, липопротеины, окисленные фосфолипиды и окисленные липопротеиды низкой плотности (ЛПНП) также запускают передачу сигналов TLR2 и TLR4 в различных типах клеток, включая моноциты/макрофаги, что приводит к активации их сигнальных путей. Хотя воспалительный ответ, вызванный ожирением, запускается преимущественно в адипоцитах и иммунных клетках, вполне вероятно, что воспалительные изменения развиваются и непосредственно в миокардиоцитах. Производство провоспалительных цитокинов в миокарде, включая такие, как фактор некроза опухоли альфа (TNFα), интерлейкины 1 и 6 (IL-1, IL-6), инициируется в том числе и из-за выраженного накопления в везикулах миокардиоцитов богатых триглицеридами остатков липопротеинов и всех вышеописанных липидных токсических метаболитов.
Развитие реакций неспецифического воспаления в миокарде и инфильтрация миокардиоцитов воспалительными клетками способствуют дополнительной продукции АФК и изменению потока трансмембранного кальция, тем самым приводя к нарушению процессов расслабления миокарда уже на ранней стадии ЛК [18]. На более поздней стадии ЛК происходит активация ренин-ангиотензин-альдостероновой системы, а накопившийся избыток токсичных метаболитов стимулирует трансформирующий фактор роста β1. Тот, в свою очередь, запускает дифференцировку фибробластов в миофибробласты, способствуя выработке коллагена и блокируя секрецию ингибиторов протеаз, вследствие чего снижается деградация внеклеточного матрикса посредством активации как канонических, так и неканонических сигнальных путей [8]. Эти воспалительные сигнальные пути способствуют развитию и прогрессированию фиброза миокарда, дисфункции митохондрий, стрессу эндоплазматического ретикулума, гибели клеток и появлению систолической и диастолической дисфункции с прогрессией в сердечную недостаточность [19].
ПОТЕНЦИАЛЬНЫЕ ВАРИАНТЫ ТЕРАПЕВТИЧЕСКОЙ СТРАТЕГИИ ПРИ ЛИПОТОКСИЧЕСКОЙ КАРДИОМИОПАТИИ
Известно, что ранняя стадия диастолической дисфункции и ГЛЖ обратимы, что подчеркивает важность профилактических вмешательств для предотвращения развития ЛК и сердечной недостаточности [20]. Однако в настоящее время отсутствуют данные крупных клинических исследований с первичными конечными точками у пациентов с ЛК. Следует также помнить, что, хотя это состояние может протекать бессимптомно, больные с ЛК и развившейся симптоматической сердечной недостаточностью должны получать терапию как пациенты с хронической сердечной недостаточностью без ЛК в соответствии с национальными клиническими рекомендациями.
Таким образом, оптимальной стратегией лечения и профилактики ЛК, а также развития СНсФВ служит целенаправленное и комплексное воздействие на все факторы риска, особенно патогенетические, такие как инсулинорезистентность, висцеральное ожирение, дислипидемия или СД 2. Традиционно все мероприятия с лечебной или профилактической направленностью делятся на немедикаментозные и медикаментозные, которые далее и будут подробно обсуждаться.
НЕМЕДИКАМЕНТОЗНЫЕ МЕРЫ
Ожирение – фактор риска развития сердечной недостаточности, при этом его распространенность значительно выше среди пациентов с СНсФВ, чем среди больных сердечной недостаточностью со сниженной фракцией выброса (СНснФВ) [2]. Хотя ожирение не ассоциируется с риском неблагоприятных исходов у пациентов с установленной СНснФВ (в чем заключается недавно описанный «парадокс ожирения»), тем не менее клиническое исследование показало значимое увеличение риска смерти у больных с висцеральным ожирением и СНсФВ [21]. Таким образом, полученные результаты указывают на необходимость снижения массы тела в подобных клинических ситуациях с целью улучшения прогноза.
Рекомендации по снижению массы тела обычно включают призывы к увеличению ежедневной физической активности, а также улучшению рациона питания за счет перераспределения энергетической составляющей ежедневного рациона в пользу более здоровых продуктов. В исследованиях было показано, что физическая активность и изменения в рационе питания служат стандартными вмешательствами для контроля над массой тела, которые также способствуют улучшению состояния у пациентов с МС, ССЗ и снижают преждевременную смертность [22].
Известно, что рафинированные углеводы, равно как и некоторые компоненты жиров, включая трансжиры и насыщенные жирные кислоты животного происхождения, способствуют развитию ожирения, СД 2, инсулинорезистентности, ССЗ и онкологических заболеваний, поскольку являются весьма токсичными для организма веществами [14]. Поэтому крайне важно донести до пациента информацию не столько о принципе снижения общего потребление калорий ежедневно, сколько об отказе от употребления этих токсичных питательных веществ в целях профилактики развития не только ЛК, но и других распространенных заболеваний современности [22].
АНТИДИАБЕТИЧЕСКАЯ ТЕРАПИЯ
Эпидемиологические данные показали, что увеличение гликированного гемоглобина (HbA1c) на каждый 1% связано с повышением риска сердечной недостаточности на 8%, а снижение на 1% связано со снижением риска на 21% для любых сердечно-сосудистых осложнений, связанных с СД 2, включая смерть [23]. Таким образом, вполне можно было бы ожидать, что снижение гипергликемии с помощью антидиабетических препаратов, уменьшающих выраженность инсулинорезистентности, может уменьшить и риск развития ЛК.
Однако в отличие от известного положительного эффекта от терапии АГ клиническая польза от гликемического контроля при установленном диагнозе сердечной недостаточности до сих пор не определена [2]. Более того, было показано, что интенсивный гликемический контроль не только не полезен, но и может быть даже вредным, приводя к эпизодам гипогликемии, которые играют роль триггеров для развития таких состояний, как аритмия, сердечная недостаточность, острый коронарный синдром и внезапная сердечная смерть. Также на сегодняшний день известно, что многие классы антидиабетических препаратов, включая сульфонилмочевину, тиазолидиндионы и ингибиторы дипептидилпептидазы-4, не приносят существенной пользы пациентам с СД 2 и сердечной недостаточностью и даже могут в некоторых случаях увеличивать частоту госпитализаций по поводу сердечной недостаточности [23].
Таким образом, эти данные позволяют предположить, что при сердечной недостаточности нацеливание только на снижение уровня глюкозы в крови недостаточно для уменьшения макрососудистых осложнений. Однако за последнее время ситуация несколько изменилась в лучшую сторону: недавние клинические исследования ряда групп антидиабетических средств продемонстрировали некоторые положительные их эффекты в отношении сердечной недостаточности. Так, агонисты рецептора глюкагоноподобного пептида-1 (аГЛП-1) лираглутид и семаглутид в исследованиях LEADER и SUSTAIN-6 соответственно показали благоприятные результаты с точки зрения сердечно-сосудистых исходов у пациентов с СД 2 [24].
В свою очередь, при приеме ингибиторов натрий-глюкозного котранспортера 2 (иНГЛТ 2), в частности эмпаглифлозина, были получены наиболее благоприятные результаты с точки зрения снижения сердечно-сосудистых конечных точек и риска госпитализаций по поводу сердечной недостаточности у пациентов с СД 2 и высоким риском ССЗ [25]. В дальнейшем благоприятное влияние иНГЛТ 2 на госпитализацию и смертность от сердечной недостаточности было также подтверждено в реальной клинической практике крупным многонациональным современным анализом [26].
Тем не менее важно отметить, что иНГЛТ2 и аГЛП-1 не смогли снизить частоту осложнений и госпитализации по поводу сердечной недостаточности у пациентов с СД 2 без атеросклеротического ССЗ, что свидетельствует об отсутствии или минимальном положительном влиянии этих современных классов антидиабетических средств на развитие сердечной недостаточности при ЛК [27].
Необходимо подчеркнуть чрезвычайную важность дальнейших исследований в данном направлении. Они необходимы, чтобы продемонстрировать и объяснить основные механизмы, с помощью которых аГЛП-1 и иНГЛТ 2 улучшают сердечно-сосудистые события при СД 2 и атеросклеротических ССЗ. Также такие исследования, возможно, позволят определить, способны ли эти препараты снижать развитие или прогрессирование ЛК у пациентов с ожирением или инсулинорезистентностью в отсутствие СД 2.
ГИПОЛИПИДЕМИЧЕСКАЯ ТЕРАПИЯ
Дислипидемия часто является сопутствующей патологией у пациентов с ожирением, инсулинорезистентностью или СД 2. Высокий уровень ЛПНП служит общепризнанным фактором риска развития атеросклеротических ССЗ. Интенсивное снижение ЛПНП с помощью статинов показано в качестве первичной и вторичной профилактики ССЗ и смертности от них, особенно у пациентов с повышенным генетическим риском.
Однако есть два факта, которые необходимо помнить в ситуации, когда речь идет о ЛК: во-первых, терапия статинами увеличивает частоту развития инсулинорезистентности и СД 2, а во-вторых, польза статинов в снижении сердечной смертности не была установлена для пациентов с хронической сердечной недостаточностью [28, 29].
Дислипидемия у пациентов с ожирением, инсулинорезистентностью и СД 2 также характеризуется повышенным уровнем триглицеридов (ТГ) и низким содержанием холестерина липопротеидов высокой плотности (ЛПВП). Данные недавних эпидемиологических исследований позволяют предположить, что гипертриглицеридемия независимо ассоциирована с повышенным риском ССЗ, возможно, из-за чрезмерного транспорта жирных кислот в миокардиоциты и повышенной вследствие этого продукции провоспалительных цитокинов [30].
Учитывая, что чрезмерное накопление внутриклеточных липидов и активация воспаления относятся к отличительным признакам ЛК, эти данные предполагают потенциальную выгоду от снижения уровня ТГ в контексте профилактики ЛК. Европейское общество кардиологов (ESC) совместно с Европейской ассоциацией по вопросам СД (EASD) в 2019 г. выпустили клинические рекомендации по профилактике и лечению ССЗ у пациентов с СД 2 и предиабетом [31]. В них пациентам с высоким уровнем ТГ (≥2,3 ммоль/л (200 мг/дл)) рекомендовано сделать главными целями терапии модификацию образа жизни: в первую очередь речь об усилении контроля над весом тела, ограничении приема алкоголя, а также стремлении к контролю за гликемией.
Добавление фибратов к терапии статинами рекомендовано только у пациентов с одновременным повышением ТГ и снижением ЛПВП, поскольку в исследованиях была показана польза от такой терапии лишь в указанной ситуации. В случае недостижения контроля за уровнем ТГ с помощью статинов или фибратов могут использоваться высокие дозы омега-3-полиненасыщенных жирных кислот (4 г/сут) [31].
ПОТЕНЦИАЛ ВЛИЯНИЯ СОВРЕМЕННОЙ ТЕРАПИИ НА ИЗМЕНЕННЫЙ СУБСТРАТНЫЙ МЕТАБОЛИЗМ МИОКАРДИОЦИТОВ
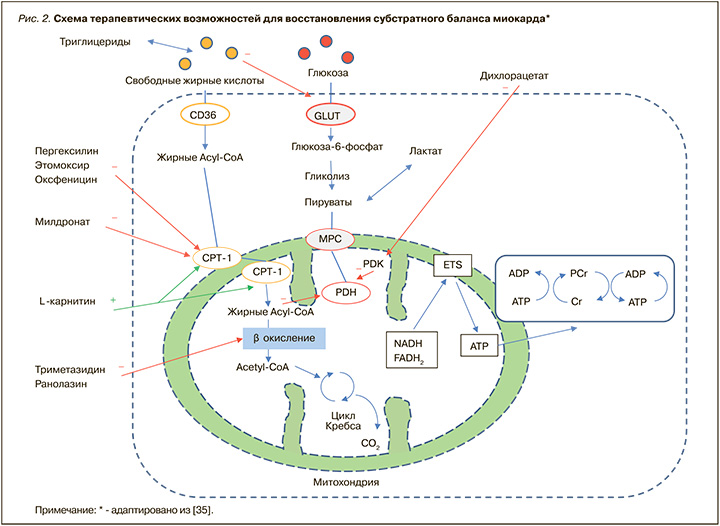
Несмотря на то что связь между развитием ЛК и состояниями, обусловленными процессами инсулинорезистентности, достаточно тесная, до настоящего времени все терапевтические стратегии, основанные на применении препаратов, контролирующих гипергликемию, инсулинорезистентность и гиперлипидемию, не привели к уменьшению риска развития СНсФВ [32]. Следовательно, существует потребность в поиске новых эффективных терапевтических путей для снижения распространенности и частоты развития как ЛК, так и сопряженной с ней сердечной недостаточности. Таким образом, влияние препаратов, оказывающих модулирующее действие на субстратный метаболизм миокардиоцитов, может стать новой терапевтической стратегией в улучшении состояния миокардиальной функции. На рисунке 2 представлен механизм действия нескольких групп препаратов, применяющихся для восстановления субстратного баланса миокарда.
Следует отметить, что разработка лекарственных средств, успешно корректирующих метаболизм миокардиоцитов, ведется достаточно давно. В настоящее время во всех странах мира с успехом применяются препараты этого класса, которые, в зависимости от их места воздействия в миокардиоците, можно разделить на несколько групп.
Первая группа самая многочисленная, ее представители оказывают блокирующее действие на ключевые ферменты-переносчики жирных кислот, лимитирующие поступление субстрата: карнитин-пальмитоил трансферазу-1 (КПТ-1) и карнитин-пальмитоил трансферазу-2 (КПТ-2). К этой группе относятся пергексилин, этомоксир, оксфеницин и мельдоний. Необходимо отметить, что, кроме мельдония, ни один из перечисленных препаратов не представлен в России и не доступен для клинического применения в отличие от стран Европы, США и Канады.
Вторую группу составляют препараты, ингибирующие финальную реакцию в процессе β-окисления жирных кислот: триметазидин и ранолазин, третью – дихлорацетат, активирующий пируватдегидрогеназы (он тоже не зарегистрирован в России).
Следует подчеркнуть, что существенные различия в механизмах лечебного воздействия между представителями трех указанных групп значимо влияют на их клинический потенциал и перспективы применения при различных патологиях. Принимая во внимание, что возникновение избыточного накопления липидов и их дальнейшего токсического воздействия на миокардиоциты выступает ключевым механизмом развития ЛК и концентрического ремоделирования левого желудочка [33], логично предположить, что устранение несоответствия между уровнями поступления и утилизации жирных кислот явилось бы оптимальным профилактическим и лечебным воздействием на описываемую патологию.
Поскольку представители второй группы средств, корректирующих метаболизм миокардиоцитов (ранолазин и триметазидин), ингибируют лишь последнюю реакцию β-окисления жирных кислот, их действие не предотвращает ни избыточное поступление липидов, ни дальнейшее накопление токсичных метаболитов в миокарде. Поэтому их эффективность ограничена атеросклеротическими ССЗ без влияния на развитие ЛК и связанную с ней сердечную недостаточность. Проведенные клинические исследования подтвердили отсутствие профилактического эффекта у представителей этой группы в отношении развития сердечной недостаточности у пациентов с СД 2 [34].
В противоположность второй группе практически все представители первой группы средств, корректирующих метаболизм миокардиоцитов (мельдоний и др.), подтвердили свою эффективность у пациентов с различным патогенезом сердечной недостаточности: они продемонстрировали улучшение сократительной функции миокарда, уменьшение симптомов и снижение класса сердечной недостаточности, а также улучшение кардиального метаболизма и диастолической функции [34]. Учитывая, что механизм их действия, а именно ограничение поступления и утилизации жирных кислот за счет блокирования КПТ-1 (для мельдония, пергексилина, этомоксира и оксфеницина) и КПТ-2 (только для мельдония), нацелен на ключевое звено в патогенезе ЛК и кардиального стеатоза, включение таких препаратов в комплексную терапию можно считать оптимальным вариантом профилактики и лечения ЛК и ее осложнений. Поскольку же среди них лишь мельдоний обладает как общими для группы преимуществами, так и спектром дополнительных благоприятных эффектов, о нем следует сказать подробнее.
Мельдоний (оригинальный препарат Милдронат®) был разработан в середине 1970-х гг. в Институте органического синтеза Академии наук Латвийской ССР группой ученых во главе с профессором И. Калвиньшем, а в1984 г. также был запатентован в США [35, 36]. На сегодняшний день в России мельдоний включен в перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения как препарат для лечения заболеваний сердца [37].
Милдронат® по своей структуре является конкурентным ингибитором γ-бутиробетаингидроксилазы – фермента, превращающего эндогенный γ-бутиробетаин в карнитин. Милдронат® также обладает способностью уменьшать транспорт карнитина из мест синтеза и абсорбции благодаря конкурентному воздействию на специфический белок-транспортер OCTN2 (organic carnitine cation transporter 2). Таким образом, в основе одного из его терапевтических эффектов лежит уменьшение содержания свободного карнитина в плазме, что приводит к уменьшению карнитин-зависимого β-окисления жирных кислот вследствие блокирования карнитин-зависимых транспортных систем. Это делает Милдронат® универсальным и высокоэффективным регулятором субстратного метаболизма миокарда (см. рис. 2) [38]. Так, с одной стороны, под действием Милдроната происходит выраженное ограничение транспорта жирных кислот в митохондрии, что предотвращает чрезмерное накопление токсичных липидных метаболитов, развитие кардиального стеатоза и ЛК. С другой стороны, при его применении происходит существенное снижение интенсивности β-окисления жирных кислот, т.е. Милдронат® объединяет в своем механизме действия все терапевтические эффекты представителей как первой, так и второй групп средств, корректирующих метаболизм миокардиоцитов.
Однако это далеко не все терапевтические возможности Милдроната, ведь параллельно с регуляторным метаболическим воздействием препарат реализует еще целый спектр полезных эффектов. Так, вслед за уменьшением концентрации карнитина происходит увеличение синтеза его предшественника γ-бутиробетаина, обладающего свойствами индуктора оксида азота – наиболее эффективного из эндогенных антиоксидантов и эндотелиопротекторов. Благодаря этому становятся возможными такие эффекты Милдроната, как улучшение микроциркуляции, снижение периферического сосудистого сопротивления, уменьшение вызванных норадреналином или ангиотензином II спазмов кровеносных сосудов, торможение агрегации тромбоцитов и увеличение эластичности мембран эритроцитов. За счет комплекса терапевтических эффектов Милдронат® оказывает селективное действие на зону ишемии, не влияя на не затронутые ишемией участки, т.е. не вызывает «эффект обкрадывания» [38]. В клинических исследованиях у пациентов с ИБС и сердечной недостаточностью, в том числе на фоне ожирения, инсулинорезистентности и СД 2, терапия Милдронатом способствовала [39–48]:
- повышению сократимости миокарда;
- увеличению толерантности к физической нагрузке;
- снижению функционального класса сердечной недостаточности;
- урежению частоты приступов стенокардии;
- улучшению показателей липидного и углеводного обмена;
- повышению качества жизни пациентов.
С целью уточнения всех достоверных клинических преимуществ Милдроната у пациентов с сердечной недостаточностью нами также был предпринят библиографический поиск в базах данных Pubmed. com, Elibrary.ru, в Центральной научной медицинской библиотеке и в Российской государственной библиотеке. Найденные публикации оценивались с использованием таких критериев, как уровень достоверности доказательств (УДД) и уровень убедительности рекомендаций (УУР) в соответствии с требованиями оценочных шкал, отраженных в приказе Минздрава России от 22.02.2019 № 103н. Результаты поиска и обработки данных позволили сделать следующие практические выводы:
1. У пациентов с хронической сердечной недостаточностью при включении в базисную терапию препарата Милдронат® в дозе 500–1500 мг/ сут курсом от 4 нед до 3 мес происходит достоверное повышение толерантности к физической нагрузке. Препарат способствует нормализации показателей гемодинамики, параметров ЭКГ и ЭхоКГ, а также повышает качество жизни пациентов (УДД 2, УУР A).
2. На фоне терапии Милдронатом в дозе 500–1500 мг/сут на протяжении от 4 нед до 3 мес наблюдается значимое улучшение периферического кровотока и внутрисердечной гемодинамики у пациентов с хронической сердечной недостаточностью (УДД 2, УУР B).
3. При использовании Милдроната достоверно снижается интенсивность перекисного окисления липидов и проявления окислительного стресса, также отмечено благоприятное влияние препарата на липидный обмен (УДД 2, УУР C).
ЗАКЛЮЧЕНИЕ
Таким образом, эффективность и безопасность препарата Милдронат® в терапии сердечной недостаточности подтверждена клиническими исследованиями. Спектр благоприятных терапевтических эффектов Милдроната, нацеленных на основные патогенетические дефекты в развитии ЛК, свидетельствует о целесообразности его применения в составе комплексной терапии сердечной недостаточности, в том числе у пациентов с инсулинорезистентностью, висцеральным ожирением и СД 2.


