
Первичный склерозирующий холангит (ПСХ, стенозирующий холангит, семейный крупноочаговый склероз) – это хроническое, медленно прогрессирующее холестатическое заболевание печени неизвестной этиологии, характеризующееся негнойным деструктивным воспалением и фиброзированием внутри- и внепеченочных желчных протоков с появлением участков их стенозирования, облитерации и исходом во вторичный билиарный цирроз печени [1, 2, 3].
ПСХ чаще болеют мужчины (2:1). Истинную распространенность заболевания в связи с трудностями диагностики установить сложно. Как правило, болезнь возникает в возрасте 25–45 лет [1, 2].
Этиология ПСХ не установлена. Большинство авторов рассматривают его как аутоиммунное заболевание [1, 2, 3].
У половины больных ПСХ распознают на бессимптомной стадии. К типичным признакам заболевания относятся зуд, боль в правом подреберье, слабость, потерю массы тела, эпизоды лихорадки. Реже болезнь манифестирует на стадии цирроза и осложнений портальной гипертензии. Самый частый биохимический маркер ПСХ – повышение уровня щелочной фосфатазы (ЩФ), нередко наблюдается также повышение уровня трансаминаз [1, 3].
Перекрестным, или overlap-синдромом в медицине принято называть состояние, которое характеризуется проявлением симптомов нескольких аутоиммунных заболеваний у одного пациента, при котором происходит взаимное отягощение двух и более болезней [4]. Подобные перекрестные синдромы встречаются и при ПСХ. У 70–80% больных ПСХ сочетается с хроническими воспалительными заболеваниями кишечника (ВЗК), чаще с язвенным колитом (ЯК) (60–70%). Изолированное течение ПСХ наблюдается у 25–30% больных [4, 5]. Необходимость раннего выявления, тщательного наблюдения и активного лечения пациентов с сочетанием данных нозологий определяется повышенным риском развития колоректального рака [1, 2, 5]. Данных о сочетании мембранозной нефропатии и ПСХ нами в литературе найдено не было. Гломерулонефрит можно отнести к редким внекишечным проявлениям ЯК. В литературе можно найти описание мембранозной нефропатии (МН), ассоциированной с семейным ЯК, у 12-летней девочки, а также случай МН у 69-летней женщины (ЯК был диагностирован спустя 3 года после дебюта нефротического синдрома). В опубликованной в 2014 г. работе O. Warling и соавт. указано, что авторы нашли лишь 6 случаев МН при ЯК. Кроме того, высказано предположение о возможной роли рецептора фосфолипазы А2 в патогенезе МН, ассоциированной с ЯК [6].
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
Пациент М., 35 лет, европеоидная раса, уроженец Сургута, инженер, впервые обратился за медицинской помощью в 2007 г. в возрасте 24 лет, когда на фоне полного благополучия отметил появление кожного зуда и сыпи в виде красных пятен на коже кистей. Проходил обследование, терапию (местно) у аллерголога, дерматолога. Верифицирована мокнущая экзема. После лечения была отмечена положительная динамика в виде купирования кожных проявлений. Эпизодически пациента беспокоил кратковременный кожный зуд, купирующийся самостоятельно.
В 2012 г. в ходе медицинского осмотра у больного были выявлены повышение аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ) более 10N, гипербилирубинемия (общий билирубин 30,2 мкмоль/л) за счет конъюгированной фракции. Впервые верифицированные цитолитический синдром и синдром холестаза послужили причиной госпитализации в стационар, где в гастроэнтерологическом отделении был проведен дифференциально-диагностический поиск между гепатитами различной этиологии. Маркеров вирусных гепатитов, антимитохондриальных антител, равно как и иных изменений по результатам проведенного лабораторно-инструментального обследования, обнаружено не было. Выставлен диагноз «хронический холестатический гепатит высокой степени активности». На фоне терапии урсодезоксихолевой кислотой (УДХК) отмечена положительная динамика в виде нормализации лабораторных показателей.
В марте 2013 г. при прохождении профилактического осмотра у пациента вновь выявлено повышение трансаминаз. При непрямой эластометрии печени зарегистрирован фиброз F3. Проведен онкопоиск, дифференциальный диагноз с гемобластозами, убедительных критериев вышеназванных заболеваний обнаружено не было. По результатам ультразвукового обследования обнаружена умеренная лимфаденопатия (подмышечные, подчелюстные, паховые лимфатические узлы max до 3 см), гепатомегалия (правая доля 176 мм, левая доля 93 мм). От проведения фиброколоноскопии в указанную госпитализацию пациент категорически отказался. При обследовании установлены следующие лабораторные показатели: АЛТ – 313 U/l, АСТ – 195 U/l, гамма-глутамилтрансфераза (ГГТ) – 863 U/l, ЩФ – 491 U/l. По результатам магнитно-резонансной томографии (МРТ) органов брюшной полости обнаружены признаки гепатита (умеренная гепатомегалия, перипортальный фиброз). Проведена пункционная биопсия печени, гистологическое исследование: хронический гепатит с минимальной активностью процесса (индекс гистологической активности 2), с фиброзом и расширением портальных трактов. Продолжена прежняя терапия желчегонными средствами, гепатопротекторами с положительным эффектом.
В 2014 г. пациент был вновь госпитализирован с клинико-лабораторными признаками холестаза. Проведена МРТ органов брюшной полости: выявлены гепатоспленомегалия, картина портальной гипертензии, холецистохолангита. Учитывая анамнестические данные, впервые выставлен диагноз «первичный склерозирующий холангит», назначена регулярная терапия УДХК.
В июле 2015 г. больной отметил ухудшение состояния в виде выраженной прогрессирующей общей слабости, появления слизи и крови в кале. По данным фиброколоноскопии, на которую пациент впервые дал согласие, обнаружены признаки язвенного панколита минимальной степени активности (слизистая оболочка отечная, со множеством микроабсцессов и налетом фибрина, без контактной кровоточивости). Выставлен диагноз «язвенный колит, тотальное поражение, впервые выявленный». Назначена терапия препаратом 5-аминосалициловой кислоты с положительным эффектом в виде купирования симптомов.
Несмотря на кажущееся стабильное, медикаментозно контролируемое течение, заболевание прогрессировало. В октябре 2015 г. у пациента впервые появились периорбитальные отеки, отеки стоп с постепенным нарастанием до анасарки, дестабилизация артериального давления с подъемами до 180/100 мм рт.ст. Больной был госпитализирован в нефрологическое отделение. В ходе обследования обнаружены критерии нефротического синдрома (протеинурия до 9 г/сут, гипопротеинемия до 37 г/л, гипоальбуминемия до 18 г/л, гиперхолестеринемия до 14,4ммоль/л), впервые зарегистрирована азотемия (креатинин до 294 мкмоль/л, мочевина до 14 мкмоль/л, скорость клубочковой фильтрации 25 мл/мин/1,73м2), обнаружены перинуклеарные антинейтрофильные цитоплазматические антитела (pANCA). С учетом прогрессирования заболевания, полисимптомности клинической картины был вновь проведен дифференциально-диагностический поиск между онкопатологией (в том числе гемобластозами), системными васкулитами, диффузными заболеваниями соединительной ткани. Убедительных критериев указанных нозологий обнаружено не было.
Была проведена пункционная нефробиопсия. Начата инициальная иммуносупрессивная системная терапия кортикостероидами, антипротеинурическая терапия ингибиторами ангиотензипревращающего фермента (иАПФ), антиагрегантами. От гиполипидемической терапии статинами решено было воздержаться, учитывая наличие ПСХ со стойким повышением трансаминаз. При снижении дозировки в 2016 г. до 25 мг преднизолона был зарегистрирован гиповолемический криз. В связи с прогрессирующим ухудшением состояния пациент самостоятельно обратился за медицинской помощью и был обследован в клинике EPMA, Brussels, EU (снимок 1–5), а также в клинике Первого Санкт-Петербургского государственного медицинского университета им. акад. И.П. Павлова, где проходил лечение с диагнозом «нефротический синдром – вторичная мембранозная нефропатия». Была продолжена терапия кортикостероидами в инициальных дозировках, начата терапия циклофосфамидом; с учетом высокого риска прогрессирования почечной недостаточности спустя 6 мес по достижении ремиссии инфузии циклофосфамида были прекращены. Пациент регулярно принимал кортикостероиды в поддерживающей дозировке, препараты 5-аминосалициловой кислоты, желчегонные и гепатопротекторы, иАПФ, ингибиторы протонной помпы, препараты кальция и витамина D, антиагреганты.
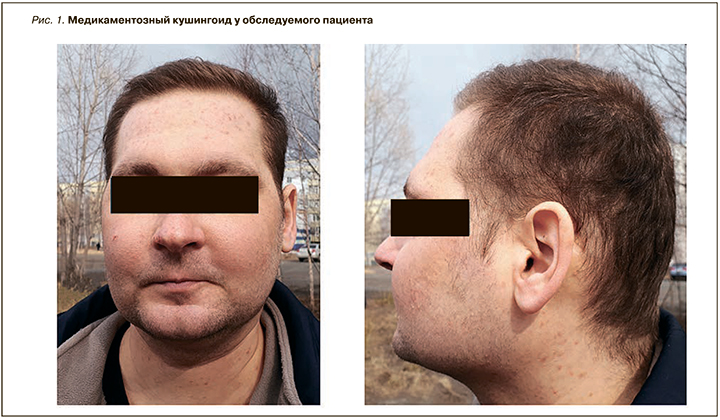
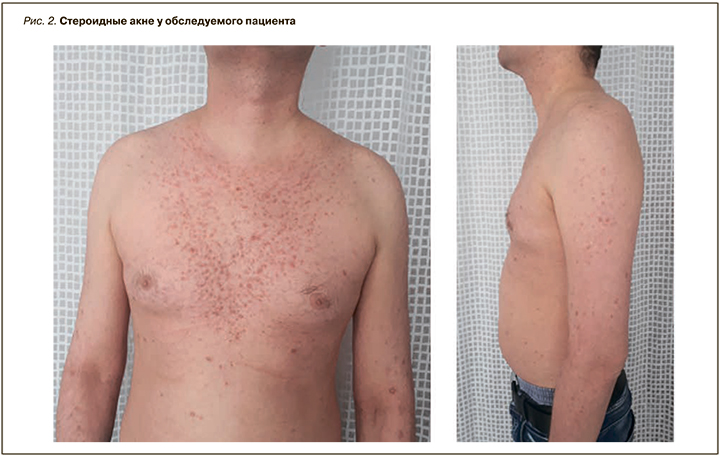
Рецидив МН у пациента был зарегистрирован в декабре 2017 г. при очередном снижении дозы кортикостероидов до 25 мг. Больной был госпитализирован в стационар. При объективном обследовании обращали на себя внимание медикаментозный кушингоид (рис. 1), сухость и бледность кожного покрова, слизистых оболочек, снижение тургора кожи, стероидные акне на коже груди, спины, конечностей (рис. 2), периорбитальные отеки, отеки стоп, нижних третей голеней, гепатомегалия (размеры печени 16–11–10 (+4)).
Из-за резистентности больного к терапии кортикостероидами и алкилирующими средствами начата терапия препаратом анти-CD20 (ритуксимаб). Однако во время очередной госпитализации в 2018 г. в ходе обследования у пациента сохранялись холестаз, нефротический синдром, признаки активности ЯК: АЛТ 95,0 U/l, АСТ 68,0 U/l, ЩФ 340,0 U/l, ГГТ 1241 U/l, общий белок 35 г/л, альбумин 13 г/л, протеинурия 6 г/сут, асцит, гепатомегалия (правая доля 160 мм, левая – 94 мм по результатам ультразвукового исследования), кашицеобразный стул до 5 раз в сутки с примесью неизмененной крови до 5 мл, кожный зуд.
Пациенту выставлен диагноз «OVERLAP-синдром: первичный склерозирующий холангит в сочетании с язвенным колитом, хроническое рецидивирующее течение, тотальное поражение, хронический гломерулонефрит (вторичная мембранозная нефропатия, pANCA-pos.), нефротический синдром. ХБП С1А4. Симптоматическая артериальная гипертензия, медикаментозно достигнутая 1 степень, риск 4 (очень высокий). ХСН 0. Хронический панкреатит с внешнесекреторной недостаточностью».
На фоне проведения антицитокиновой терапии (регулярные инфузии ритуксимаба) удалось снизить дозировку преднизолона до 10 мг без признаков рецидива заболеваний.
ОБСУЖДЕНИЕ
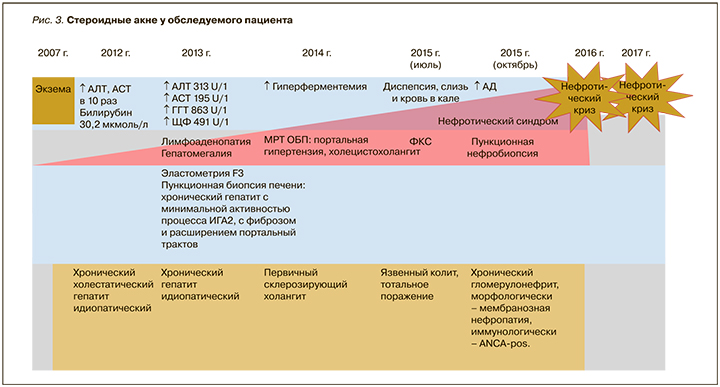
Подводя итог (рис. 3), необходимо отметить важные особенности данного случая.
Пол и возраст пациента соответствуют литературным данным. Среди больных с сочетанием ПСХ и ЯК преобладают мужчины, чаще всего диагноз устанавливается в возрасте 30–40 лет [2]. Именно эти факторы определяют агрессивность течения и резистентность к терапии.
У 15–55 % больных ПСХ клинические симптомы могут длительное время отсутствовать. У большинства из них отмечается медленное развитие ПСХ, однако уже за много лет до установления диагноза больные жалуются на нарастающую утомляемость. Одной из наиболее частых жалоб у 60–90% пациентов является мучительный, изнуряющий кожный зуд, в связи с чем на коже можно обнаружить многочисленные расчесы. Возникновение кожного зуда связывают с холемией и центральными механизмами [2]. Не исключено, что именно этим можно объяснить первые клинические проявления нашего пациента в 2007 г., расцененные специалистами как мокнущая экзема.
Вполне закономерно обнаружение у мужчины и ЯК. В настоящее время доказанной является связь между воспалительными заболеваниями кишечника и аутоиммунными поражениями гепатобилиарной системы. Частота ПСХ у больных ЯК составляет в среднем 2–26% [2]. При этом наличие сопутствующего поражения печени зависит от протяженности воспалительного процесса в кишечнике (в данном клиническом наблюдении речь идет о панколите): если при дистальных формах язвенного колита ПСХ встречается у 0,5% пациентов, то в случае распространенного поражения кишечника сочетание с ПСХ достигает 5%. Что касается клинической картины, то течение ПСХ в сочетании с воспалительными заболеваниями кишечника не отличается от такового у пациентов с изолированным ПСХ (что мы наблюдаем у пациента М.); течение же ЯК при сочетании с ПСХ носит более мягкий характер [2].
Регистрация ЯК, ПСХ «в перекресте» с почечной патологией в литературе освещено крайне скудно. Известно, что одним из вариантов поражения почек выступает МН, носящая вторичный характер. У взрослых она служит самой частой причиной нефротического синдрома, достигая 20–40%, протекая тяжелее у мужчин, чем у женщин [7]. В связи с этим рассмотренный клинический случай, несомненно, представляет крайний интерес.
Необходимо отметить и сложность терапии наблюдаемого пациента. Принципы терапии данных заболеваний, учитывая общность их этиопатогенетических механизмов, сходны. Однако неэффективность проводимой иммуносупрессивной терапии объясняется высокой активностью и агрессивным взаимоотягощающим течением заболеваний (ЯК, ПСХ, МН). Согласно стратификации риска, факторами неблагоприятного прогноза в отношении прогрессирования МН в данном случае являются мужской пол, артериальная гипертензия, гистологические признаки тубулоинтерстициального фиброза и канальцевой атрофии, суточная протеинурия нефротического уровня. Тяжесть течения ПСХ определяют молодой возраст, мужской пол, гипербилирубинемия, гистологическая картина биоптата печени. Мужской пол, молодой возраст, манифестация заболевания с появления крови в кале, результаты гистологического исследования биоптата с обнаружением микроабсцессов, панколит являются критериями агрессивного течения ЯК. Вероятно, наличие данных факторов у пациентов должно способствовать более раннему началу антицитокиновой терапии.
В целом стоит отметить классическое течение заболеваний. Учитывая наличие характерных клинических проявлений, а также результаты проведенного исследования, диагноз «OVERLAP-синдром» не вызывает сомнений.
ЗАКЛЮЧЕНИЕ
Редкое сочетание нозологический форм в виде триады заболеваний (ПСХ, ЯК, МН), ранее не нашедшее отражения в литературе, трудность верификации основного диагноза на фоне полисимптомной клинической картины, частые рецидивы и резистентность к проводимой терапии на фоне взаимоотягощающего течения заболеваний определяют интерес, актуальность и ценность данного клинического наблюдения. Общность этиопатогенетических механизмов развития и системность поражения в описанном случае напоминают привычные для нас нозологии, что приводит к затяжному периоду дифференциально-диагностического поиска и, как следствие, отсроченному активному лечению основного заболевания. На это стоит обратить внимание при проведении дифференциальной диагностики патологических состояний, сопровождающихся холестатическим, а также нефротическим синдромами у мужчин молодого возраста.


