
ВВЕДЕНИЕ
Ревматоидный артрит (РА) – системное, хроническое иммуновоспалительное заболевание неизвестной этиологии, которое сопровождается патологией суставов и имеет различные внесуставные проявления, включая поражение органов сердечно-сосудистой системы [1, 2]. Глобальная распространенность РА оценивается в 0,46% населения всего мира (в некоторых регионах от 0,06 до 1,27%), в России же она составляет 0,61%. Заболевание может дебютировать в любом возрасте, но чаще поражает лиц трудоспособного возраста и в 2–3 раза чаще встречается у женщин, чем у мужчин [3, 4].
Согласно данным обзора 50 исследований (n=91 618 пациентов), основной причиной смерти у пациентов с РА выступали кардиоваскулярные заболевания (33 250 случаев смерти – 39,6% от общей выборки) [5]. В двух крупных метаанализах c участием более 150 000 пациентов РА был связан с повышением риска сердечно-сосудистых событий (ССС) на 48% (относительный риск (ОР) 1,48; 95% доверительный интервал (ДИ): 1,36–1,62) и на 50% более частым возникновением сердечно-сосудистых заболеваний (ССЗ) [6, 7].
Вероятность развития ССЗ у пациентов с РА в 1,5–2 раза выше, чем у лиц того же возраста и пола из общей популяции. Этот повышенный риск объясняется, кроме традиционных факторов риска, высоким системным хроническим воспалением, которое является признаком РА (рис. 1) [8].
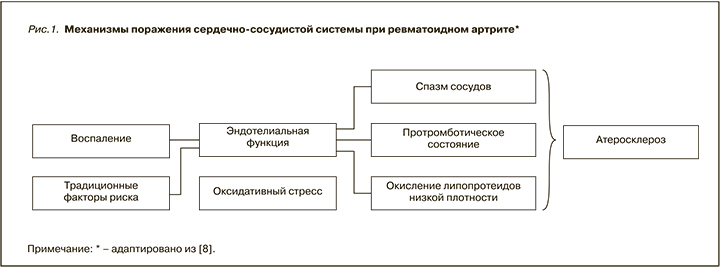
Расчет факторов риска сердечно-сосудистых осложнений может способствовать их раннему выявлению и вмешательству, особенно в популяции высокого риска, такой как пациенты с РА. Однако калькуляторы, разработанные для населения в целом, включая шкалу Framingham и алгоритм SCORE, склонны к недооценке риска кардиоваскулярных осложнений у пациентов с РА [9, 10], и лишь британский QRISK2 до недавнего времени [11] включал встроенный параметр РА как фактор, влияющий на совокупную оценку риска. Для решения этой проблемы в рекомендациях рабочей группы Европейской антиревматической лиги (EULAR) по лечению РА от 2017 г. для шкалы SCORE рекомендуется использовать коэффициент умножения 1,5 в моделях прогнозирования риска сердечно-сосудистых осложнений для всех пациентов с РА без поправки на длительность заболевания и внесуставные проявления, как это было ранее [12]. Кроме того, для оценки индивидуального риска кардиоваскулярных осложнений у пациентов с РА были разработаны различные инструменты, включая расширенный рейтинг риска (ERS-RA), калькулятор трансатлантического сердечно-сосудистого риска (ATACC-RA), шкалы ASSIGN и QRISK3 [13, 14].
РОЛЬ ТРОМБОЦИТОВ В АУТОИММУННЫХ РЕАКЦИЯХ И ВОСПАЛЕНИИ ПРИ РЕВМАТОИДНОМ АРТРИТЕ
Тромбоциты снабжены кластерами дифференцировки мегакариоцитов, различными поверхностными рецепторами и гликопротеинами, элементами цитоскелета, гранулами и гладкой трубчатой системой эндоплазматического ретикулума [15]. Эти разнообразные свойства вместе с повсеместным присутствием тромбоцитов делают их идеальными кандидатами на роль иммунных клеток. Многими исследователями тромбоциты рассматриваются в качестве активных регуляторов врожденного и адаптивного иммунитета [16]. Поскольку хроническое системное воспаление рассматривается как ось патогенеза аутоиммунных заболеваний и служит причиной повышенного риска ССС, исследование вклада тромбоцитов в эти взаимодействия представляется особенно важным [17, 18].
Являясь продуктом мегакариоцитопоэза, тромбоциты представляют собой ануклеарные клетки с продолжительностью жизни 8–10 дней [19]. В течение этого относительно короткого периода, особенно в первые 24 ч, они могут синтезировать белки на своей мРНК и производить микрочастицы [20]. Известно также, что тромбоциты вырабатывают большое количество трансформирующего фактора роста бета (ТФР-β), который подавляет чрезмерную активацию тромбоцитов и разрушение соединительной ткани, но может и не оказывать своего благотворного действия при РА в силу нескольких причин, наиболее вероятные из которых – полиморфизмы в некоторых генах [21, 22].
Помимо участия в гемостазе, активация тромбоцитов приводит к высвобождению различных цитокинов и хемокинов, которые обеспечивают привлечение лейкоцитов и их адгезию к поврежденному эндотелию. Интерлейкин-1β (ИЛ-1β), полученный из тромбоцитов, синтезируется при активации эндотелиальных клеток. Инкубация этих клеток в присутствии тромбин-активированных тромбоцитов индуцирует зависимую от ИЛ-1β секрецию ИЛ-6, ИЛ-8, CCL-2 (моноцитарного хемотаксического фактора-1 – MCH-1) и увеличивает экспрессию молекул адгезии ICAM-1 и α v β 3 за счет эндотелиальных клеток [23, 24]. Кроме того, происходящий из тромбоцитов ИЛ-1β инициирует активацию NF-κB в эндотелиоцитах и тем самым индуцирует NF-κB-зависимую транскрипцию гена хемокинов [25].
CD40L (CD154) является секретируемой молекулой, заслуживающей особого внимания в контексте аутоиммунитета. Этот мембранный гликопротеин относится к членам семейства фактора некроза опухолей-альфа (ФНО-α) и экспрессируется активированными тромбоцитами [26]. В течение короткого периода, длящегося от нескольких минут до нескольких часов, поверхностно экспрессируемый CD40L расщепляется и высвобождается в растворимой форме (sCD40L). Считается, что более 95% sCD40L имеет тромбоцитарное происхождение и действует как триггер для активации эндотелия, увеличивая экспрессию рецепторов воспалительной адгезии (VCAM-1 и ICAM-1), вызывая выработку хемокинов (CCL-2, ИЛ-6 и ИЛ-8) и стимулирование выработки матриксной металлопротеиназы-9 (ММП-9) [15, 27]. Связывание sCD40L с CD40 на эндотелиальных клетках вызывает высвобождение медиаторов, привлекающих лейкоциты [28]. sCD40L участвует в регуляции функции Т-клеток, активации дендритных клеток и регуляции Т-зависимого переключения изотипа антител; он также обеспечивает новый механизм аутоактивации тромбоцитов и образования гомотипических агрегатов тромбоцитов [29]. Более того, sCD40L служит прогностическим биомаркером ССС, таких как острый инфаркт миокарда (ОИМ) и острое нарушение мозгового кровообращения (ОНМК), а его уровень у пациентов с РА коррелирует с уровнями ревматоидного фактора (РФ) IgM и IgG [29].
Существует большое количество хемокинов и цитокинов, выделяемых из альфа-гранул тромбоцитов, которые учувствуют в формировании и поддержании воспаления [30] (табл.).
Повышение количества и активности тромбоцитов у пациентов с РА может приводить к снижению их продолжительности жизни, тем самым способствуя более быстрому обновлению пула клеток c преобладанием молодых тромбоцитов, которые отличаются увеличенными размерами и повышенной реактивностью [32]. Повышенное количество растворимых рилизатов тромбоцитов, таких как Р-селектин, бета-тромбоглобулин или тромбоцитарный фактор роста 4, обнаруживается в сыворотке пациентов с РА наряду с большим количеством циркулирующих PMP (микрочастиц тромбоцитов) [33]. Повышенная активация тромбоцитов, оцениваемая по этим показателям, положительно коррелирует с тяжестью течения РА [34, 35].
Известно, что антитела к циклическому цитруллинированному пептиду (АЦЦП) и РФ могут активировать тромбоциты. Иммунные комплексы АЦЦП-IgE стимулируют тромбоциты через FccRIIA и FceRIa – рецепторы с высоким и низким сродством (FceRII/CD23) для IgE и рецептор низкоаффинного IgG (FcγRIIa) [36]. Процент АЦЦП к общему IgG показывает положительную корреляцию с активацией тромбоцитов и активностью РА [37]. Наличие этих антител [38, 39] было предложено как новый фактор риска ССС [40, 41] и нестабильности атеросклеротических бляшек [42, 43].
У пациентов с РА тромбоциты связываются с экспонированным коллагеном через мембранный рецептор гликопротеина VI (GPVI), и это взаимодействие провоцирует генерацию PMP и ИЛ-1. Повышенное количество PMP обнаруживается не только в плазме, но и синовиальной жидкости. Фактически концентрация PMP в синовиальной жидкости значительно выше, чем в крови; это позволяет предположить, что его избыточное образование происходит локально [44].
ИЛ-1 служит ключевой молекулой для взаимодействия между тромбоцитами и фибробластоподобными синовиоцитами (ФС) [45]. Активированные ФС считаются ключевыми эффекторами разрушения хряща [46] и дефектного ангиогенеза при РА [47]. По сравнению со здоровыми лицами, ФС пациентов с РА экспрессируют измененные уровни цитокинов, хемокинов, молекул адгезии и ММП, которые устойчивы к апоптозу. Их взаимодействие с тромбоцитами приводит к повышению продукции простагландина I2 (ПГ I2). PMP стимулируют повышенный синтез эйкозаноидов – циклооксигеназы 2-го типа (ЦОГ-2), микросомального простагландина Е-синтазы 1 (mPGES-1) и ПГ E2 [48].
PMP экспрессируют антигены тромбоцитов и связываются с другими аутоантигенами, присутствующими в плазме [49]. Антигены, происходящие из тромбоцитов, включают внутриклеточные белки, такие как виментин. Экстернализация виментина происходит во время активации тромбоцитов. Поверхность PMP может быть местом дальнейших посттранскрипционных модификаций аутоантигенов, например цитруллинизации [50]. Это приводит к образованию неоэпитопов, распознаваемых аутоантителами, характерными для РА [51]. Компоненты комплемента (C1q, C3 и C4) также могут связываться с поверхностью PMP и обнаруживаться в синовиальной жидкости пациентов с РА. Было показано, что эти иммунные комплексы активируют нейтрофилы, в результате чего самоподдерживающаяся активация тромбоцитов и нейтрофилов в месте синовита может участвовать в образовании цитруллинированного фибриногена и неоэпитопов виментина [50]. Белки тромбоцитарного происхождения трансформируются пептидиларгининдезиминазой 4 (PAD4) – ферментом, полученным из нейтрофилов [52]. Активация тромбоцитов и нейтрофилов вызывает нетоз, который, в свою очередь, активирует ФС, способные интернализировать нейтрофильные внеклеточные ловушки. Также происходит индуцирование свойств антигенпрезентирующих клеток в ФС, которые могут представлять полученные из тромбоцитов, посттранскрипционно модифицированные аутоантигены [53, 54].
Наконец, тромбоциты способствуют развитию синовита при РА, поддерживая постоянную проницаемость синовиального микроциркуляторного русла. Они активируют эндотелиальные клетки, что влечет за собой экспрессию молекул поверхностной адгезии и способствует миграции клеток [55]. Тромбоциты выделяют серотонин в местах повреждения сосудов, способствуя образованию эндотелиальных щелей субмикронного размера и синовиальной инфильтрации воспалительными клетками [56].
ВЛИЯНИЕ ТРОМБОЦИТОВ НА СЕРДЕЧНО-СОСУДИСТУЮ ПАТОЛОГИЮ У ПАЦИЕНТОВ С РЕВМАТОИДНЫМ АРТРИТОМ
Некоторые исследования показали повышение количества тромбоцитов и тромбоцитарных факторов роста в синовии и синовиальной жидкости, ассоциированное с увеличением РФ [57]. При РА активированные тромбоциты, отдельно или вместе с другими воспалительными медиаторами, могут играть важную роль в тромбообразовании, нарушении синовиальной микроциркуляции и разрушении хряща. Тромбоциты взаимодействуют с эндотелиальными клетками и лейкоцитами в синовиальных сосудах при воспалении. Последующая агрегация тромбоцитов приводит к образованию тромбов и изменению синовиальной микроциркуляции [58]. P-селектин – молекула адгезии, вырабатываемая тромбоцитами и эндотелиальными клетками, способствует взаимодействию тромбоцитов, лейкоцитов и эндотелиальных клеток при воспалении [59]. Другие белки, продуцируемые тромбоцитами, также проявляют провоспалительное действие, которое может приводить к повреждению суставов. Так, тромбоцитарный фактор роста вызывает синовит и паннусоподобную гиперплазию в экспериментальной модели РА [60].
Повышенный уровень среднего объема тромбоцитов (MPV) связан с высоким сердечно-сосудистым риском у пациентов с артериальной гипертензией, ожирением, сахарным диабетом 2-го типа, курением и гиперхолестеринемией, а также с таким ССС, как дестабилизация атеросклеротической бляшки, нестабильная стенокардия, ОИМ и пароксизмальная форма фибрилляции предсердий [61–63]. MPV рассматривается как независимый фактор риска и предиктор ОИМ у лиц с высоким кардиоваскулярным риском [64, 65]. В большой проспективной когорте пациентов с установленным цереброваскулярным заболеванием (n=3134) увеличенный уровень MPV выступал предиктором ОНМК в течение 4 лет наблюдения. При этом увеличение MPV на каждый фемтолитр ассоциировалось с возрастанием ОР ОНМК на 11% [66]. В другом исследовании было выявлено значительное увеличение уровня MPV у пациентов с семейной средиземноморской лихорадкой, что указывает на связь между воспалением, активацией тромбоцитов и протромботическим состоянием [67].
Тем не менее были и попытки связать один маркер функции тромбоцитов (например, Р-селектин, MPV) с ускоренным атеросклерозом и ССЗ при РА, но они не увенчались успехом [68].
Относительно простой тест – агрегатометрия, являющаяся «золотым стандартом» для определения функции тромбоцитов. Она может быть выполнена с использованием обогащенной тромбоцитами плазмы или цельной крови и различных агонистов тромбоцитов (например, кислоты, аденозиндифосфата, коллагена, тромбина, эпинефрина и модифицированных иммуноглобулинов). Агонист добавляется в суспензию, после чего регистрируется динамический показатель агрегации тромбоцитов. Будучи тестом in vitro, агрегатометрия имеет ограничения (например, отсутствие взаимодействия с другими клетками крови, артефакты, возникающие во время забора образцов, центрифугирования и разделения тромбоцитов). Агрегатометрия на основе подсчета количества тромбоцитов в цельной крови преодолевает некоторые из этих ограничений. Некоторые исследования доказали, что агрегатометрия тромбоцитов полезна для выявления гиперактивности тромбоцитов при РА [69] и оценки эффективности противоревматических препаратов [70, 71]. Исследования агрегатометрии тромбоцитов выявили триггеры чрезмерной агрегации тромбоцитов: это серопозитивность по РФ, наличие антител к бета-2-микроглобулинам и циркулирующие иммунные комплексы [72, 73]. Интересно, что повышенная чувствительность тромбоцитов in vitro к агонистам и аутоиммунным факторам была обнаружена и при других ревматических заболеваниях (например, при ревматической полимиалгии, системном склерозе, подагре), где общие механизмы образования иммунных комплексов и ускоренного атерогенеза могут быть подавлены препаратами с антиагрегантам эффектом. Тромбоцитоз (>400×109/л), характерный для активного РА, был ассоциирован с повышенной чувствительностью к агонистам тромбоцитов, таким как коллаген и эпинефрин, что позволяет предположить наличие патологической связи между тромбоцитозом, тромбоцитами [74] и каскадом арахидоновой кислоты [75].
ОСОБЕННОСТИ ТРОМБОЦИТОПОЭЗА У ПАЦИЕНТОВ С РЕВМАТОИДНЫМ АРТРИТОМ
Распространенность тромбоцитоза при РА неизвестна, однако уровень тромбоцитов и их параметры могут коррелировать с активностью заболевания, а также вносить свой вклад в развитие ССЗ. В одном из исследований в Саудовской Аравии, в которое вошли 140 пациентов с РА, встречаемость тромбоцитоза составила 16% [76]. Точный патогенетический механизм, вызывающий увеличение количества тромбоцитов при РА, до сих пор не до конца понятен [77]. В литературе имеются данные о том, что провоспалительные плейотропные цитокины, участвующие в патогенезе РА, обладают мегакариоцитопоэтическими и/или тромбопоэтическими свойствами. Более того, несколько гемопоэтических цитокинов с доминантным клонированием могут также действовать как реагенты острой фазы и способствовать развитию воспаления [77]. Факторы роста, способствующие развитию и поддержанию воспаления при РА, а именно ИЛ-1, ИЛ-3, ИЛ-4, ИЛ-6, ИЛ-11, ФНО-α, фактор стволовых клеток, фактор ингибирования лейкемии, гранулоцитарный колониестимулирующий фактор, тромбопоэтин (ТПО) и эритропоэтин (ЭПО), принимают участие в мегакариоцитопоэзе во время активного воспалительного процесса. Некоторые данные указывают, что ТПО может способствовать реактивному тромбоцитозу при РА. В семействе цитокинов gp130, не являющихся клон-специфическими, ИЛ-6, по-видимому, преобладает в индукции мегакариопоэза. Однако другие цитокины и факторы роста также могут вносить вклад в патологический мегакариоцитопоэз при РА (рис. 2) [78].
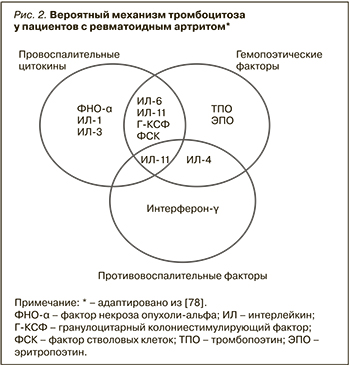
В исследовании Kiraz S. et al. пациенты с РА и нормальным уровнем тромбоцитов имели значения ТПО, сравнимые с группой контроля. Концентрации ТПО у пациентов с легким тромбоцитозом (450–650×109/л) были значительно повышены, тогда как у пациентов с уровнем тромбоцитов тромбоцитов более 650×109 снижены. Таким образом, уровень ТПО может быть связан с реактивным тромбоцитозом у пациентов с РА высокой активности [79].
РОЛЬ ТРОМБОЦИТОЗА В ТЕЧЕНИИ РЕВМАТОИДНОГО АРТРИТА
В исследовании Hutchinson R. et al. было продемонстрировано, что у пациентов с РА наблюдается взаимосвязь между количеством тромбоцитов и активностью заболевания. Также было отмечено, что повышение уровня тромбоцитов коррелировало с тяжестью анемии, сидеропенией, лейкоцитозом и уровнем РФ по сравнению с больными РА, имевшими нормотромбоцитемию (p <0,001). Более того, у пациентов с тромбоцитозом системные проявления РА (поражения легких, ревматоидные узелки, кожный васкулит) встречались чаще, однако данная связь не была значимой (p >0,05) [80].
Схожие данные были получены в исследовании Schmitt-Sody M. et al., где была выявлена корреляция между уровнем тромбоцитов и активностью РА, лейкоцитозом, повышенным уровнем РФ, а также скоростью оседания эритроцитов (СОЭ). Кроме того, была дана оценка положительному влиянию растворимого Р-селектина на адгезию лейкоцитов к эндотелиоцитам [58].
В другом исследовании, выполненном Ertenli I. et al., было установлено, что пациенты с РА и тромбоцитозом, по сравнению с пациентами без повышения тромбоцитов, имели более высокую активность заболевания, повышенные уровни СОЭ и С-реактивного белка (СРБ), растворимого Р-селектина и большую продолжительность утренней скованности [81]. Farr M. et al., кроме взаимосвязи тромбоцитоза с клинико-лабораторной активностью, обнаружили снижение продолжительности жизни тромбоцитов у пациентов с РА высокой активности и гипертромбоцитозом при сохранении нормальной агрегационной способности тромбоцитов [82].
Несколько коллективов авторов в серии описания случаев показали, что результатом тромбоцитоза, высокой адгезии и агрегации тромбоцитов могут быть рецидивирующие тромбоэмболии различной локализации. При этом риск развития гиперкоагуляции и тромбоэмолических осложнений во всех случаях напрямую зависел от активности течения РА [83–85]. При анализе цитокинового спектра у пациентов с РА и гипертромбоцитозом было выявлено повышение уровней ИЛ-1β, ИЛ-4 и ИЛ-6, тогда как у больных с нормальным или сниженным уровнем тромбоцитов концентрация этих цитокинов не возрастала [77, 86]. При оценке влияния уровня тромбоцитов на поражение суставов Kacena M. et al. установили, что пациенты с тромбоцитозом имели более высокую рентгенологическую стадию поражения суставов за одинаковый период болезни. Несмотря на участие мегакариоцитов в регуляции процессов синтеза костной ткани, провоспалительные цитокины, по всей видимости, подавляют эту активность, усиливая, в свою очередь, остеокластогенез и приводя к формированию эрозий [87].
ВЛИЯНИЕ ТРОМБОЦИТАРНЫХ ИНДЕКСОВ НА АКТИВНОСТЬ РЕВМАТОИДНОГО АРТРИТА
Различные авторы изучали связь между активностью РА и уровнями гемоглобина, количеством тромбоцитов и MPV. В исследовании Talukdar M. et al. были оценены все эти параметры и их влияние на активность РА у 80 пациентов, из которых 48 пациентов имели высокую (DAS28 >5,1) и 32 – умеренную или низкую (DAS 28 <5,1) степени активности заболевания. Пациенты с высокой активностью имели значительно более высокий уровень тромбоцитов и MPV, но более низкий уровень гемоглобина, чем пациенты со средней и низкой активностью, как среди мужчин, так и женщин (р <0,001) [88]. Этот результат согласуется с выводами, сделанными в предыдущих исследованиях, проведенных Milovanovic M. et al. [86], Yazici S. et al. [89], где так же были выявлены положительные взаимосвязи между уровнем MPV и активностью РА. В то же время в исследовании Moghimi J. et al. не было обнаружено существенной разницы между уровнями MPV у пациентов с различной активностью РА [90]. Напротив, Kisacik B. et al. [91] и Gasparyan A. et al. [92] продемонстрировали снижение MPV у пациентов с активным РА.
Khaled S. et al. была предложена модель оценки активности РА при помощи показателей тромбоцитов [93]. Основную группу этого исследования составили 98 пациентов с РА; в группу контроля вошли 53 человека без РА, которые были сопоставимы с основной по возрасту и полу. В качестве исследуемых параметров были выбраны СОЭ, СРБ, индексы тромбоцитов из общего анализа крови (количество тромбоцитов (PLT), MPV, ширина распределения тромбоцитов (PDW) и тромбокрит (PCT). Ввиду того что MPV, PDW, СРБ и СОЭ были выше у пациентов с РА, на основе этих параметров был разработан новый индекс, получивший название New DAS: EgyDAS = 5,78 + 0,65 × ln(СОЭ) + 0,37 × ln(СРБ) – 7,47/√PDW – 3,09 × ln(MPV). Этот индекс коррелировал с DAS28. Кроме того, новая шкала продемонстрировала возможности идентификации пациентов с РА (пороговое значение <-0,79) и их стратификации в соответствии с активностью заболевания (низкая, средняя, высокая или ремиссия) [93].
ЗАКЛЮЧЕНИЕ
В настоящее время не вызывает сомнений тот факт, что пациенты с иммуновоспалительными заболеваниями, включая РА, имеют повышенные риски развития ССЗ. При этом известные механизмы и доказанные факторы риска ССЗ не полностью объясняют повышенную сердечно-сосудистую смертность у данной когорты пациентов. Имеющиеся данные свидетельствуют о том, что тромбоциты могут выступать факторами прогрессирования ССЗ, участвуют в воспалении, дисфункции эндотелия, тромбоза и являются потенциальными мишенями для противоревматоидной и сердечно-сосудистой терапии при РА.
Системное воспаление, опосредованное многочисленными цитокинами (ФНО-α, ИЛ-6, ИЛ-8), факторами роста и аутоантителами, стимулирует оборот тромбоцитов в костном мозге при РА. Тромбоциты могут превосходить лейкоциты, моноциты и другие клетки в производстве Р-селектина, CD40L, тромбоцитарного фактора роста и таким образом занимать одну из лидирующих позиций в процессе системного воспаления. Тромбоциты, тромбоцитарный фактор 4, тромбоцитарные факторы роста, серотонин и микрочастицы были обнаружены в синовиальной жидкости пациентов с РА; там эти агенты могут нарушать микроциркуляцию и поддерживать воспаление. Однако весьма вероятно, что циркулирующие тромбоциты, включая те, что происходят из синовиальной жидкости, обладают более важной васкулопатической функцией. Поэтому важно дальнейшее изучение количества, функции, роли тромбоцитов и тромбоцитопоэза у пациентов с РА различной активности и их динамики на фоне применения базисной и генно-инженерной биологической терапии.


